





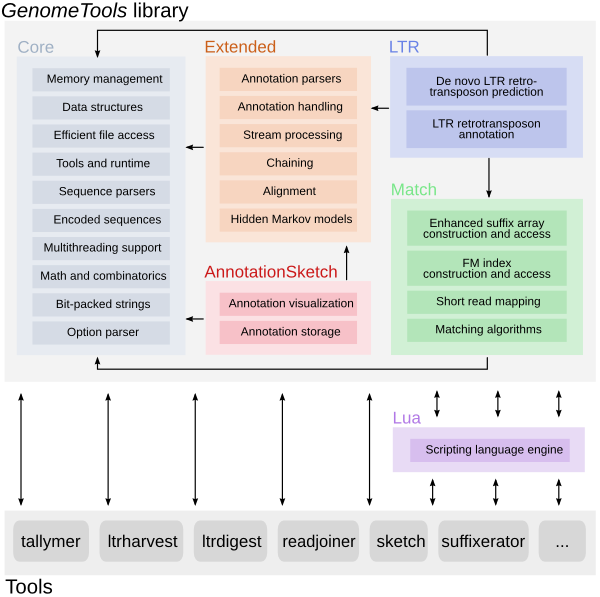
The GenomeTools genome analysis system is a free collection of bioinformatics
tools (in the realm of genome informatics) combined into a single binary named
gt. It is based on a C library named libgenometools which contains a wide
variety of classes for efficient and convenient implementation of sequence and
annotation processing software.
If you are interested in gene prediction, have a look at GenomeThreader.
GenomeTools has been designed to run on every POSIX compliant UNIX system, for example, Linux, macOS, and OpenBSD.
Debian and Ubuntu users can install the most recent stable version simply using apt, e.g.
apt-get install genometools(as root) to install the gt executable. To install the library and development headers, use
apt-get install libgenometools0 libgenometools0-devinstead. This is not required to just use the tools.
If Homebrew is installed, GenomeTools can be installed on
supported macOS versions using brew:
brew install genometoolsTo use GenomeTools on systems that do not have native packages, or to modify GenomeTools at build time, you need to build from source. Source tarballs are available from GitHub. For instructions on how to build the source by yourself, have a look at the INSTALL file. In most cases (e.g. on a 64-bit Linux system) something like
make -j4should suffice. On 32-bit systems, add the 32bit=yes option. Add cairo=no if
you do not have the Cairo libraries and their development headers installed.
This will, however, remove AnnotationSketch support from the resulting binary.
When your binary has been built, use the install target and prefix option to
install the compiled binary on your system. Make sure you repeat all the options
from the original make run. So
make -j4 install prefix=~/gtwould install the software in the gt subdirectory in the current user's home
directory. If no prefix option is given, the software will be installed
system-wide (requires root access).
GenomeTools uses a collective code construction contract for contributions (and the process explains how to submit a patch). Basically, just fork this repository on GitHub, start hacking on your own feature branch and submit a pull request when you are ready. Our recommended coding style is explained in the developer's guide (among other technical guidelines).
To report a bug, ask a question, or suggest new features, use the GenomeTools issue tracker.








































































































































