![]()
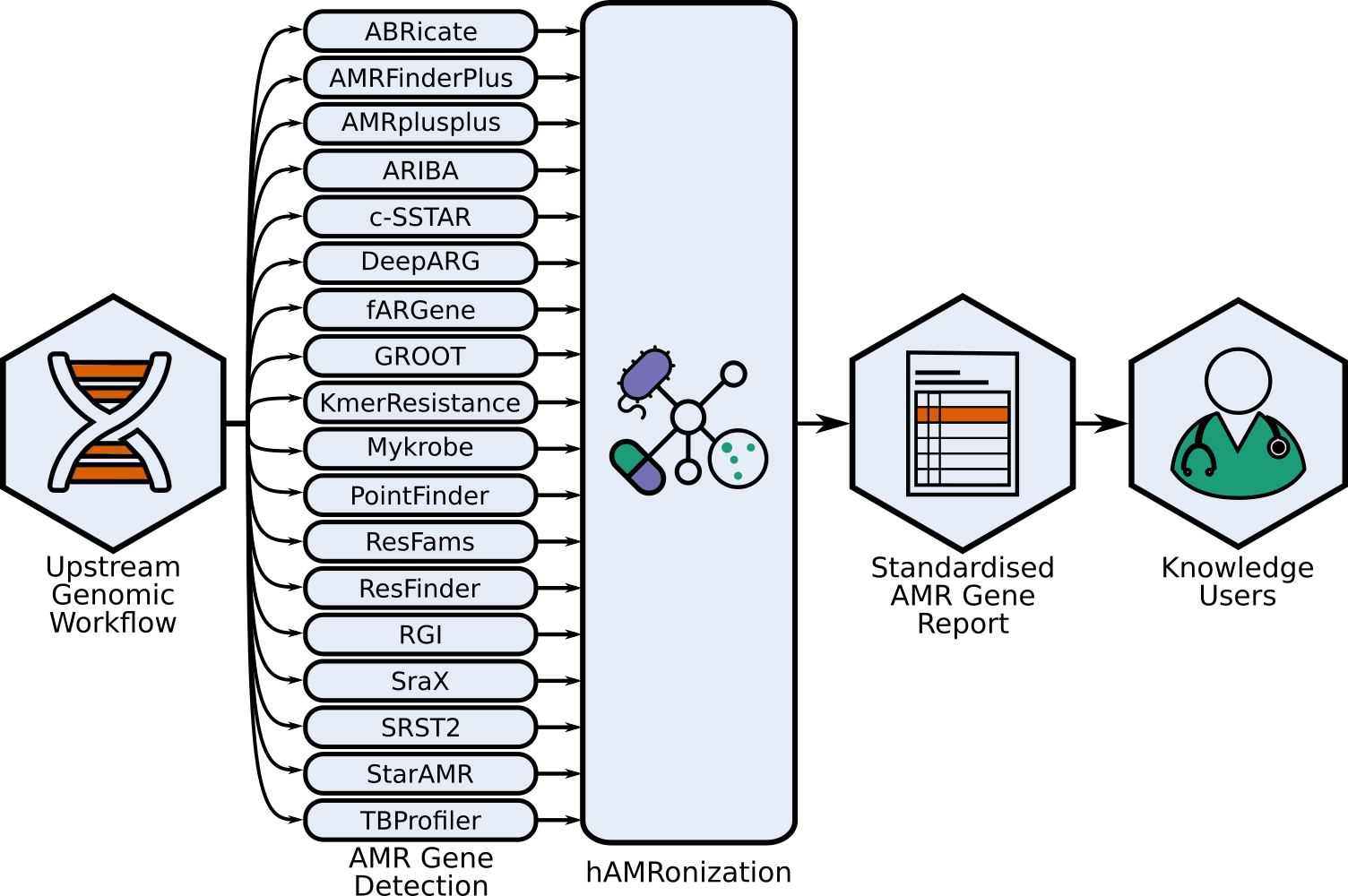
This repo contains the hAMRonization module and CLI parser tools combine the outputs of 18 (as of 2022-09-25) disparate antimicrobial resistance gene detection tools into a single unified format.
This is an implementation of the hAMRonization AMR detection specification scheme which supports gene presence/absence resistance and mutational resistance (if supported by the underlying tool).
This supports a variety of summary options including an interactive summary.
This tool requires python>=3.7 and pandas and the latest release can be installed directly from pip, conda, docker, this repository, or from the galaxy toolshed:
pip install hAMRonization

Or
conda create --name hamronization --channel conda-forge --channel bioconda --channel defaults hamronization



Or to install using docker:
docker pull finlaymaguire/hamronization:latest
Or to install the latest development version:
git clone https://github.com/pha4ge/hAMRonization
pip install hAMRonization
Alternatively, hAMRonization can also be installed and used in galaxy via the galaxy toolshed.
NOTE: Only the output format used in the "last updated" version of the AMR prediction tool has been tested for accuracy. Older tool versions or updates which lead to a change in output format may not work. In theory, this should only be a problem with major version changes but not all tools follow semantic versioning. If you encounter any issues with newer tool versions then please create an issue in this repository.
usage: hamronize <tool> <options>
Convert AMR gene detection tool output(s) to hAMRonization specification format
options:
-h, --help show this help message and exit
-v, --version show program's version number and exit
Tools with hAMRonizable reports:
{abricate,amrfinderplus,amrplusplus,ariba,csstar,deeparg,fargene,groot,kmerresistance,resfams,resfinder,mykrobe,pointfinder,rgi,srax,srst2,staramr,tbprofiler,summarize}
abricate hAMRonize abricate's output report i.e., OUTPUT.tsv
amrfinderplus hAMRonize amrfinderplus's output report i.e., OUTPUT.tsv
amrplusplus hAMRonize amrplusplus's output report i.e., gene.tsv
ariba hAMRonize ariba's output report i.e., OUTDIR/OUTPUT.tsv
csstar hAMRonize csstar's output report i.e., OUTPUT.tsv
deeparg hAMRonize deeparg's output report i.e.,
OUTDIR/OUTPUT.mapping.ARG
fargene hAMRonize fargene's output report i.e., retrieved-
genes-*-hmmsearched.out
groot hAMRonize groot's output report i.e., OUTPUT.tsv (from `groot
report`)
kmerresistance hAMRonize kmerresistance's output report i.e., OUTPUT.res
resfams hAMRonize resfams's output report i.e., resfams.tblout
resfinder hAMRonize resfinder's output report i.e.,
ResFinder_results_tab.txt
mykrobe hAMRonize mykrobe's output report i.e., OUTPUT.json
pointfinder hAMRonize pointfinder's output report i.e.,
PointFinder_results.txt
rgi hAMRonize rgi's output report i.e., OUTPUT.txt or
OUTPUT_bwtoutput.gene_mapping_data.txt
srax hAMRonize srax's output report i.e., sraX_detected_ARGs.tsv
srst2 hAMRonize srst2's output report i.e., OUTPUT_srst2_report.tsv
staramr hAMRonize staramr's output report i.e., resfinder.tsv
tbprofiler hAMRonize tbprofiler's output report i.e., OUTPUT.results.json
summarize Provide a list of paths to the reports you wish to summarize
To look at a specific tool e.g. abricate:
>hamronize abricate -h
usage: hamronize abricate <options>
Applies hAMRonization specification to output from abricate (OUTPUT.tsv)
positional arguments:
report Path to tool report
optional arguments:
-h, --help show this help message and exit
--format FORMAT Output format (tsv or json)
--output OUTPUT Output location
--analysis_software_version ANALYSIS_SOFTWARE_VERSION
Input string containing the analysis_software_version for abricate
--reference_database_version REFERENCE_DATABASE_VERSION
Input string containing the reference_database_version for abricate
Therefore, hAMRonizing abricates output:
hamronize abricate ../test/data/raw_outputs/abricate/report.tsv --reference_database_version 3.2.5 --analysis_software_version 1.0.0 --format json
To parse multiple reports from the same tool at once just give a list of reports as the argument, and they will be concatenated appropriately (i.e. only one header for tsv)
hamronize rgi --input_file_name rgi_report --analysis_software_version 6.0.0 --reference_database_version 3.2.5 test/data/raw_outputs/rgi/rgi.txt test/data/raw_outputs/rgibwt/Kp11_bwtoutput.gene_mapping_data.txt
You can summarize hAMRonized reports regardless of format using the 'summarize' function:
> hamronize summarize -h
usage: hamronize summarize <options> <list of reports>
Concatenate and summarize AMR detection reports
positional arguments:
hamronized_reports list of hAMRonized reports
optional arguments:
-h, --help show this help message and exit
-t {tsv,json,interactive}, --summary_type {tsv,json,interactive}
Which summary report format to generate
-o OUTPUT, --output OUTPUT
Output file path for summary
This will take a list of report and create single sorted report in the specified format just containing the unique entries across input reports. This can handle mixed json and tsv hamronized report formats.
hamronize summarize -o combined_report.tsv -t tsv abricate.json ariba.tsv
The interactive summary option will produce an html file that can be opened within the browser for navigable data exploration (feature developed with @alexmanuele).
Alternatively, hAMRonization can be used within scripts (the metadata must contain the mandatory metadata that is not included in that tool's output, this can be checked by looking at the CLI flags in hamronize <tool> --help):
import hAMRonization
metadata = {"analysis_software_version": "1.0.1", "reference_database_version": "2019-Jul-28"}
parsed_report = hAMRonization.parse("abricate_report.tsv", metadata, "abricate")
The parsed_report is then a generator that yields hAMRonized result objects from the parsed report:
for result in parsed_report:
print(result)
Alternatively, you can use the .write attribute to export all results left in the generator to a file (if a filepath isn't provided, this will write to stdout).
parsed_report.write('hAMRonized_abricate_report.tsv')
You can also output a json formatted hAMRonized report:
parsed_report.write('all_hAMRonized_abricate_report.json', output_format='json')
If you want to write multiple reports to one file, this .write method can accept append_mode=True to append rather than overwrite the output file and not include the header (in tsv format).
parsed_report.write('all_hAMRonized_abricate_report.tsv', append_mode=True)
Currently implemented parsers and the last tool version for which they have been validated:
- abricate: last updated for v1.0.0
- amrfinderplus: last updated for v3.10.40
- amrplusplus: last updated for c6b097a
- ariba: last updated for v2.14.6
- csstar: last updated for v2.1.0
- deeparg: last updated for v1.0.2
- fargene: last updated for v0.1
- groot: last updated for v1.1.2
- kmerresistance: late updated for v2.2.0
- mykrobe: last updated for v0.8.1
- pointfinder: last updated for v4.1.0
- resfams: last updated for hmmer v3.3.2
- resfinder: last updated for v4.1.0
- rgi (includes RGI-BWT) last updated for v5.2.0
- srax: last updated for v1.5
- srst2: last updated for v0.2.0
- staramr: last updated for v0.8.0
- tbprofilder: last updated for v3.0.8
The hAMRonization specification is implemented in the hAMRonizedResult dataclass.
This is a simple datastructure that uses positional and key-word args to distinguish mandatory from optional hAMRonization fields. It also uses type-hinting to validate the supplied values are of the correct type
Each parser follows a similar strategy, using a common interface.
This has been designed to match the biopython SeqIO parse function
>>> import hAMRonization
>>> filename = "abricate_report.tsv"
>>> metadata = {"analysis_software_version": "1.0.1", "reference_database_version": "2019-Jul-28"}
>>> for result in hAMRonization.parse(filename, metadata, "abricate"):
... print(result)
Where the final argument to the hAMRonization.parse command is whichever tool is being parsed.
An abstract iterator is then implemented to ingest a given AMR tool's report (via the appropriate subclassed implementation), hAMRonize results i.e. translate the original inputs to the fields in the hAMRonization specification, and yield a stream of hAMRonizedResult dataclasses.
This iterator also implements a write function to enable outputting the contents to a output stream or filehandle in either tsv or json format.
Each tool has a specific subclass of this abstract hAMRonizedResultIterator e.g. AbricateIO.AbricateIterator.
These include an attribute containing the mapping of the tools original output report fields to the hAMRonized specification fields (self.field_mapping), as well as handling specifying any additional required metadata.
The parse method of these subclasses then implements the tool-specific parsing logic required.
This is typically a simple csv.DictReader but can be more complex such as the json parsing of resfinder output,
or the modification of output fields required to better fit some tools into the hAMRonization specification.
We welcome contributions for users in any form (from github issues flagging problems/requests) to pull requests of bug fixes or adding new parsers.
First fork this repository and set up a development environment (replacing YOURUSERNAME with your github username:
git clone https://github.com/YOURUSERNAME/hAMRonization
conda create -n hAMRonization
conda activate hAMRonization
cd hAMRonization
pip install pytest flake8
pip install -e .
On every commit github actions automatically runs tests and linting to check the code. You can manually run these in your development environment as well.
To run a full set of integration tests:
pushd test
bash run_integration_test.sh
popd
To run unit tests that verify parsing validity for each tool as well as generation of valid summaries you can use pytest:
pip install pytest
pushd test
pytest
popd
Finally to run linting and check whether your code matches the project code style:
pushd hAMRonization
flake8 . --count --select=E9,F63,F7,F82 --show-source --statistics
flake8 . --count --exit-zero --max-complexity=20 --max-line-length=127 --statistics
popd
If you wish to add a parser for a new tool here are the main steps required:
-
Add an entry into
_RequiredToolMetadataand_FormatToIteratorinhAMRonziation/__init__.pywhich points to the appropriateToolNameIO.pycontaining the tool's Iterator subclass -
In
ToolNameIO.pyadd arequired_metadatalist containing any mandatory fields not implemented by the tool -
Then add a class
ToolNameIterator(hAMRonizedResultIterator)and implement the__init__methods with the approriate mapping (self.field_mapping), and metadata (self.metadata). -
To this class, add a
parsemethod which reads an opened file stream into a dictionary per line/result (matching the keys ofself.field_mapping) and yields the output ofself.hAMRonizebeing applied to that dictionary. -
To add a CLI parser for the tool, create a python file in the
parsersdirectory:from hAMRonization import Interfaces if __name__ == '__main__': Interfaces.cli_parser('toolname')
Alternatively, the hAMRonized_parser.py can be used as a common script interface to all implemented parsers.
- Finally, following the template in
test/test_parsing_validity.py, please generate a unit test that ensures the parser is working as you intend it to!
If you have any questions about any of this or need any help, please file an issue.
- What's the difference between an Antimicrobial Resistance 'Result' and 'Report'?
- For the purposes of this project, a 'Report' is an output file (or collection of files) from an AMR analysis tool. A 'Result' is a single entry in a report. For example, a single line in an abricate report file is a single Antimicrobial Resistance 'Result'.
Here are some known issues that we would welcome input on trying to solve!
-
mandatory fields:
gene_symbolandgene_nameare confusing and not usually both present (only consistently used in AFP). Means tools either need 1:2 mapping i.e. single output field maps to bothgene_symbolandgene_nameOR have fragile text splitting of single field that won't be robust to databases changes. Current solution is 1:2 mapping e.g. staramr -
inconsistent nomenclature of terms being used in specification fields: target, query, subject, reference. Need to stick to one name for sequence with which the database is being searched, and one the hit that results from that search.
-
sequence_identity: is sequence type specific %id amino acids != %id nucleotide but does this matter? -
coverage_depthseems to include both tool fields that are average depth of read and just plain overall read-count, -
contig_idisn't general enough when some tools this ID naturally corresponds to aread_name(deepARG), individual ORF (resfams), or protein sequence (AFP with protein input): change toquery_id_nameor similar?



























































































































