![]()
This repository aims to gather tools and contributors from the metabolomics world.
It is maintained by Galaxy metabolomics community and open to any contributors.
Tools themselves should stick with the IUC (Galaxy Intergalactic Utilities Commission) standards and best practices
- Bjorn Gruening (@bgruening)
- Gildas Le Corguillé (@lecorguille)
- Guitton Yann (@yguitton)
New team members can be suggested by a PR against this file which needs to be approved by a majority of the current team members.
This repository was initiated by the Workflow4Metabolomics project. Workflow4Metabolomics, W4M in short, is a collective offering ressources for processing, analyzing and annotating metabolomic data.
Galaxy is an open, web-based platform for data intensive biomedical research. Whether on the free public server or your own instance, you can perform, reproduce, and share complete analyses.
Conda is a package, dependency and environment management for any language—Python, R, Ruby, Lua, Scala, Java, JavaScript, C/ C++, FORTRAN, and more.
Planemo is a command-line utilities to assist in developing Galaxy
Some tools implement a Job Dynamic Destination Mapping, like xcmsSet
The R-package CAMERA is a Collection of Algorithms for MEtabolite pRofile Annotation. Its primary purpose is the annotation and evaluation of LC-MS data. It includes algorithms for annotation of isotope peaks, adducts and fragments in peak lists. Additional methods cluster mass signals that originate from a single metabolite, based on rules for mass differences and peak shape comparison. To use the strength of already existing programs, CAMERA is designed to interact directly with processed peak data from the R-package xcms. What it does? The CAMERA annotation procedure can be split into two parts: We want to answer the questions which peaks occur from the same molecule and secondly compute its exact mass and annotate the ion species. Therefore CAMERA annotation workflow contains following primary functions: 1. peak grouping after retention time (groupFWHM) 2. peak group verification with peakshape correlation (groupCorr) Both methods separate peaks into different groups, which we define as ”pseu- dospectra”. Those pseudospectra can consists from one up to 100 ions, de- pending on the molecules amount and ionizability. Afterwards the exposure of the ion species can be performed with: 2 1. annotation of possible isotopes (findIsotopes) 2. annotation of adducts and calculating hypothetical masses for the group (findAdducts) This workflow results in a data-frame similar to a xcms peak table, that can be easily stored in a comma separated table .csv (Excel-readable). If you have two or more conditions, it will return a diffreport result within the annotation results. The diffreport result shows the most significant differences between two sets of samples. Optionally create extracted ion chromatograms for the most significant differences.
This function check annotations of ion species with the help of a sample from opposite ion mode. As first step it searches for pseudospectra from the positive and the negative sample within a reten- tion time window. For every result the m/z differences between both samples are matched against specific rules, which are combinations from pos. and neg. ion species. As example M+H and M-H with a m/z difference of 2.014552. If two ions matches such a difference, the ion annotations are changed (previous annotation is wrong), confirmed or added. Returns the peaklist from one ion mode with recalculated annotations.
xml macros for other camera repos
xml file describing dependencies for other camera repos
This tool takes as inputs either tabular table files from the metabolomic workflow (variableMetadata, dataMatrix and sampleMetadata) or a table file of your own and can execute three different functions ("sorting", "corrdel" and "corr_matrix").
The "sorting" function: used for metabolomic workflow
- First of all, it sorts the data by pcgroup.
- It computes the mean operation of all the signal values of the metabolites by sample, and put the results in a new column "signal_moy".
- It finally creates a tabular output "sorted_variableMetadata.tsv".
The "corrdel" function: used for metabolomic workflow
For each pcgroup of the previous sorted tabular file "sorted_table.tsv", it does the following things:
- it computes a correlation matrix
- it determines the metabolites which are not correlated to others from the same pcgroup based on the threshold value filled in the "Correlation threshold for pcgroup" parameter
- the metabolites are sorted by the mean signal intensity (form the highest to the lowest), and each metabolite is tested to the previous ones in the list ; if the tested metabolite is at least correlated to one previous one, it is tagged as DEL (for "deleted", written in a column called "suppress")
It creates two additional tabular files:
- "correlation_matrix_selected.tsv" (correlation matrix of selected metabolites only)
- "sif_table.tsv" (for visualization in CytoScape, based on selected metabolites and "Cytoscape correlation threshold" filled value)
The "corr_matrix" function: used for user table file
| It computes a correlation matrix named "correlation_matrix.tsv" and creates a sif file named "sif_table.tsv" (for visualization in CytoScape).
Genform generates candidate molecular formulas from high-resolution MS data. It calculates match values (MV) that show how well candidate molecular formulas fit the MS isotope peak distributions (MS MV) and the high-resolution MS/MS fragment peak masses (MSMS MV). Finally it computes a combined match value from these two scores. This software can be regarded as a further development of the ElCoCo and MolForm modules of MOLGEN-MS with a clear specialization towards MS/MS.
Optimize free fluxes and optionaly metabolite concentrations of a given static metabolic network defined in an FTBL file to fit 13C data provided in the same FTBL file.
IPO.ipo4xcmsSet A Tool for automated Optimization of XCMS Parameters
IPO.ipo4xcmsSet A Tool for automated Optimization of XCMS Parameters
We strongly encourage you to read the documentation <https://isoplot.readthedocs.io/en/latest/>_ before using Isoplot.
Reads as set of XML-based mass-spectrometry data files and generates an MSnExp object. This function uses the functionality provided by the ‘mzR’ package to access data and meta data in ‘mzData’, ‘mzXML’ and ‘mzML’.
ASICS, based on a strong statistical theory, handles automatically the metabolite identification and quantification
BARSA is an automatic algorithm for bi-dimensional NMR spectra annotation
Spectra preprocessing
These steps correspond to the following steps in the PEPS-NMR R library (https://github.com/ManonMartin/PEPSNMR):
- Group Delay suppression (First order phase correction)
- Removal of solvent residuals signal from the FID
- Apodization to increase the Signal-to-Noise ratio of the FID
- Fourier transformation
- Zero order phase correction
- Shift referencing to calibrate the spectra with internal compound referencing
- Baseline correction
- Setting of negatives values to 0
NMR Read
Nuclear Magnetic Resonance Bruker files reading (from the PEPS-NMR R package (https://github.com/ManonMartin/PEPSNMR))
Normalization (operation applied on each (preprocessed) individual spectrum) of preprocessed data
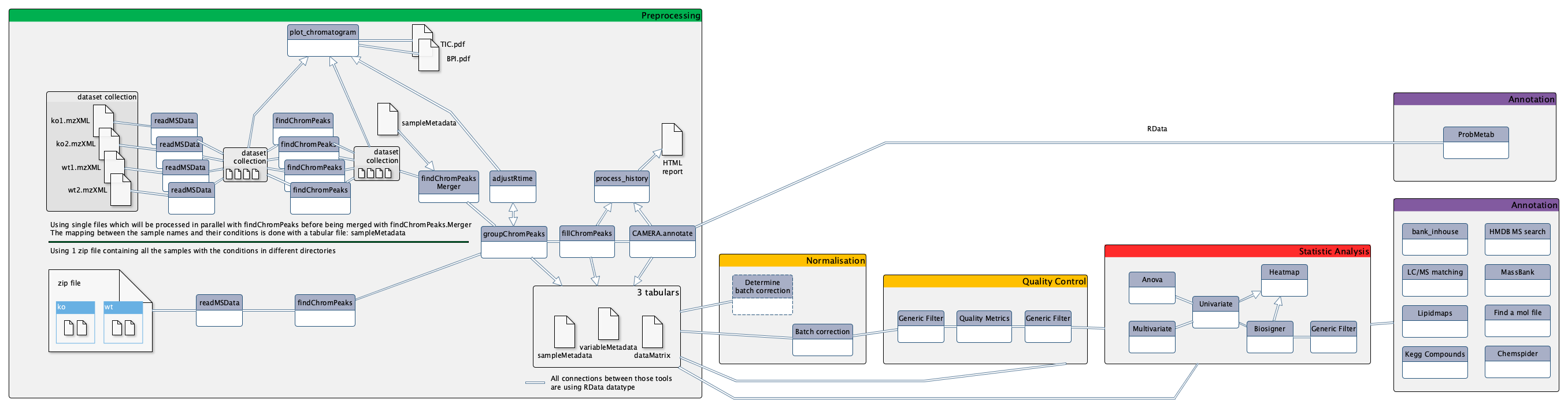
xcms get sampleMetadata This tool generates a skeleton of sampleMetadata with perhaps some strange sample names which are definitely compatible with xcms and R This sampleMetadata file have to be filled with extra information as the class, batch information and maybe conditions
xcms fillChromPeaks Integrate areas of missing peaks For each sample, identify peak groups where that sample is not represented. For each of those peak groups, integrate the signal in the region of that peak group and create a new peak.
xcms groupChromPeaks
After peak identification with xcmsSet, this tool groups the peaks which represent the same analyte across samples using overlapping m/z bins and calculation of smoothed peak distributions in chromatographic time. Allows rejection of features, which are only partially detected within the replicates of a sample class.
xml macros for other xcms repos
xcms findChromPeaks Merger This tool allows you to run one xcms findChromPeaks process per sample in parallel and then to merge all RData images into one. The result is then suitable for xcms groupChromPeaks. You can provide a sampleMetadata table to attribute phenotypic values to your samples.
xcms plot chromatogram This tool will plot Base Peak Intensity chromatogram (BPI) and Total Ion Current chromatogram (TIC) from xcms experiments.
xcms refineChromPeaks
After peak identification with xcms findChromPeaks (xcmsSet), this tool refines those peaks. It either removes peaks that are too wide or removes peaks with too low intensity or combines peaks that are too close together. Note well that refineChromPeaks methods will always remove feature definitions, because a call to this method can change or remove identified chromatographic peaks, which may be part of features. Therefore it must only be run immediately after findChromPeaks (xcmsSet).
xml file describing dependencies for other xcms repos
xcms adjustRtime After matching peaks into groups, xcms can use those groups to identify and correct correlated drifts in retention time from run to run. The aligned peaks can then be used for a second pass of peak grouping which will be more accurate than the first. The whole process can be repeated in an iterative fashion. Not all peak groups will be helpful for identifying retention time drifts. Some groups may be missing peaks from a large fraction of samples and thus provide an incomplete picture of the drift at that time point. Still others may contain multiple peaks from the same sample, which is a sign of impropper grouping.
xcms process history This tool provide a HTML summary which summarizes your analysis using the [W4M] XCMS and CAMERA tools
test data repo for xcms tool suit
xcms findChromPeaks This tool is used for preprocessing data from multiple LC/MS files (NetCDF, mzXML and mzData formats) using the xcms_ R package. It extracts ions from each sample independently, and using a statistical model, peaks are filtered and integrated. A tutorial on how to perform xcms preprocessing is available as GTN_ (Galaxy Training Network).
- Urszula Czerwinska ABiMS / IFB - CNRS/Sorbonne Université - Station Biologique de Roscoff - France
- Marion Landi PFEM / MetaboHUB - INRA - France
- Misharl Monsoor @mmonsoor - ABiMS / IFB - CNRS/Sorbonne Université - Station Biologique de Roscoff - France
- Pierre Pericard @ppericard- ABiMS / IFB - CNRS/Sorbonne Université - Station Biologique de Roscoff - France