workflow4metabolomics / tools-metabolomics Goto Github PK
View Code? Open in Web Editor NEWGalaxy tools for metabolomics maintained by Workflow4Metabolomics
Home Page: https://workflow4metabolomics.org/
License: GNU General Public License v3.0
Galaxy tools for metabolomics maintained by Workflow4Metabolomics
Home Page: https://workflow4metabolomics.org/
License: GNU General Public License v3.0
- Missing?
So dear @workflow4metabolomics/ms, (and if you have 1 minutes: @sneumann, @jotsetung) :
chrom or MSWIf you want, you can use the same checklist as me (here) and move items.
Many thanks by advanced
Example:
XSET OBJECT INFO
An "xcmsSet" object with 1 samples
Time range: Inf--Inf seconds (Inf--Inf minutes)
Mass range: Inf--Inf m/z
Peaks: 0 (about 0 per sample)
Peak Groups: 0
Sample classes: .
Optained with
ARGUMENTS INFO
singlefile_galaxyPath /export/galaxy-central/database/files/000/dataset_5.dat
singlefile_sampleName MM8.mzML
xfunction xcmsSet
xsetRdataOutput /export/galaxy-central/database/files/000/dataset_7.dat
sampleMetadataOutput /export/galaxy-central/database/files/000/dataset_8.dat
ticspdf /export/galaxy-central/database/files/000/dataset_9.dat
bicspdf /export/galaxy-central/database/files/000/dataset_10.dat
nSlaves 1
method centWave
ppm 25
peakwidth c(20, 50)
Report by Mickaël:
Since Galaxy unzip dataset, if there is only one file in it.
The xcmsSet wrapper is not design to deal with only one mzXML file
OTRS - 2016112910000118
Si on opte pour RT en minutes et qu'on veut utiliser le annotateDiffreport en tant que
variableMetadata pour faire une filtration ou qualityMetrix, ca plante car
l'identificateur d'ions est resté en seconde et du coup, il n'y a pas coh?rence entre les
fichiers dataMatrix et variablesMetadata.
Merci de votre attention
JF
Until september, we were able to manage functional tests and docker building using some wonderful Conda dependencies.
Since, there was a huge migration to R-3.3.1 bioconda/bioconda-recipes#2404
All our tools passed this migration.
But since, there are conflicts between tools versions and channels and whatever which induce that some tools come with the R 3.2.2 and some with R 3.3.1: galaxyproject/planemo#604
An update of Conda within Galaxy should solve this issue...
Currently, I'm testing as suggested here galaxyproject/tools-iuc#1071 to use the last version of miniconda : miniconda3-4.2.12
wget -q --recursive 'https://repo.continuum.io/miniconda/Miniconda3-4.2.12-Linux-x86_64.sh'
bash 'Miniconda3-4.2.12-Linux-x86_64.sh' -b -p /tmp/mc3-4.2.12/planemo conda_install --conda_prefix /tmp/mc3-4.2.12/ .
👍 Good news: all tools are installed with their 3.3.1 version
planemo test --install_galaxy --conda_dependency_resolution --conda_prefix /tmp/mc3-4.2.12/
galaxy.tools.deps.conda_util DEBUG 2016-12-21 15:53:28,567 Executing command: /tmp/mc3-4.2.12/bin/conda list --name [email protected]_1 --export > /tmp/tmpjhqeXz/tmp/jobdepsdf01NEa817a1d312c412c739d4c357c9343718c2068f458808f083afdf1f251d82564c/[email protected]_1
requests.packages.urllib3.connectionpool INFO 2016-12-21 15:53:29,172 Starting new HTTP connection (1): localhost
requests.packages.urllib3.connectionpool DEBUG 2016-12-21 15:53:29,436 "GET /api/jobs/5729865256bc2525?key=89116108df0529eaf07c60bfbc2cd985 HTTP/1.1" 200 None
galaxy.tools.deps.conda_util DEBUG 2016-12-21 15:53:29,713 Executing command: /tmp/mc3-4.2.12/bin/conda create -y --unknown --offline --prefix /tmp/tmpjhqeXz/job_working_directory/000/2/conda-env --file /tmp/tmpjhqeXz/tmp/jobdepsdf01NEa817a1d312c412c739d4c357c9343718c2068f458808f083afdf1f251d82564c/[email protected]_1 > /dev/null
requests.packages.urllib3.connectionpool INFO 2016-12-21 15:53:31,044 Starting new HTTP connection (1): localhost
requests.packages.urllib3.connectionpool DEBUG 2016-12-21 15:53:31,297 "GET /api/jobs/5729865256bc2525?key=89116108df0529eaf07c60bfbc2cd985 HTTP/1.1" 200 None
galaxy.tools.deps.conda_util DEBUG 2016-12-21 15:53:32,447 Executing command: /tmp/mc3-4.2.12/bin/conda clean --tarballs -y
galaxy.tools.deps.conda_util DEBUG 2016-12-21 15:53:33,450 Executing command: /tmp/mc3-4.2.12/bin/conda list --name [email protected] --export > /tmp/tmpjhqeXz/tmp/jobdeps1Qjc8_89b20b2c5915d075d4a0c07dfb65cfb04a50060d7fadd95d12895e582b914f79/[email protected]
requests.packages.urllib3.connectionpool INFO 2016-12-21 15:53:34,506 Starting new HTTP connection (1): localhost
galaxy.tools.deps.conda_util DEBUG 2016-12-21 15:53:34,576 Executing command: /tmp/mc3-4.2.12/bin/conda install -y --unknown --offline --prefix /tmp/tmpjhqeXz/job_working_directory/000/2/conda-env --file /tmp/tmpjhqeXz/tmp/jobdeps1Qjc8_89b20b2c5915d075d4a0c07dfb65cfb04a50060d7fadd95d12895e582b914f79/[email protected] > /dev/null
requests.packages.urllib3.connectionpool DEBUG 2016-12-21 15:53:34,717 "GET /api/jobs/5729865256bc2525?key=89116108df0529eaf07c60bfbc2cd985 HTTP/1.1" 200 None
galaxy.tools.deps.conda_util DEBUG 2016-12-21 15:53:36,610 Executing command: /tmp/mc3-4.2.12/bin/conda clean --tarballs -y
galaxy.tools.deps.conda_util DEBUG 2016-12-21 15:53:37,592 Executing command: /tmp/mc3-4.2.12/bin/conda list --name [email protected]_4 --export > /tmp/tmpjhqeXz/tmp/jobdepsHNeXMre7325fdd48bc9d36b864284fa0ff5f09769060a0bf098ef4ab24c33996193d03/[email protected]_4
galaxy.tools.deps.conda_util DEBUG 2016-12-21 15:53:38,540 Executing command: /tmp/mc3-4.2.12/bin/conda install -y --unknown --offline --prefix /tmp/tmpjhqeXz/job_working_directory/000/2/conda-env --file /tmp/tmpjhqeXz/tmp/jobdepsHNeXMre7325fdd48bc9d36b864284fa0ff5f09769060a0bf098ef4ab24c33996193d03/[email protected]_4 > /dev/null
...
UnsatisfiableError: The following specifications were found to be in conflict:
- bzip2 1.0.6 3
Use "conda info <package>" to see the dependencies for each package.
galaxy.tools.deps.conda_util DEBUG 2016-12-21 15:53:39,769 Executing command: /tmp/mc3-4.2.12/bin/conda clean --tarballs -y
galaxy.jobs.runners ERROR 2016-12-21 15:53:40,699 (2) Failure preparing job
Traceback (most recent call last):
File "/tmp/tmpjhqeXz/galaxy-dev/lib/galaxy/jobs/runners/__init__.py", line 170, in prepare_job
job_wrapper.prepare()
File "/tmp/tmpjhqeXz/galaxy-dev/lib/galaxy/jobs/__init__.py", line 913, in prepare
self.dependency_shell_commands = self.tool.build_dependency_shell_commands(job_directory=self.working_directory)
File "/tmp/tmpjhqeXz/galaxy-dev/lib/galaxy/tools/__init__.py", line 1331, in build_dependency_shell_commands
tool_instance=self
File "/tmp/tmpjhqeXz/galaxy-dev/lib/galaxy/tools/deps/__init__.py", line 104, in dependency_shell_commands
return [dependency.shell_commands(requirement) for requirement, dependency in requirement_to_dependency.items()]
File "/tmp/tmpjhqeXz/galaxy-dev/lib/galaxy/tools/deps/resolvers/conda.py", line 245, in shell_commands
self.build_environment()
File "/tmp/tmpjhqeXz/galaxy-dev/lib/galaxy/tools/deps/resolvers/conda.py", line 240, in build_environment
raise DependencyException("Conda dependency seemingly installed but failed to build job environment.")
DependencyException: Conda dependency seemingly installed but failed to build job environment.
/tmp/mc3-4.2.12/bin/conda list --name [email protected]_1 | grep bzip2
bzip2 1.0.6 3
/tmp/mc3-4.2.12/bin/conda list --name [email protected] | grep bzip2
bzip2 1.0.6 3
/tmp/mc3-4.2.12/bin/conda list --name [email protected]_4 | grep bzip2
bzip2 1.0.6 3😭
Should we propose a merge of all graphs in one or in different pages?
@yguitton - 22/02/16 to @lecorguille, @melpetera
Bonsoir
j'ai une piste enfin peut-être, pourriez-vous tester en créant deux class samples et pool pour voir?
normalement j'avais fais les modifs pour que plotTIC et plotBPC gèrent le cas à une seule classe mais bon, c'est peut-être pas parfaitdites-moi si ça règle qq chose
Yann
And example:
find: `FOO': No such file or directory
find: `2': No such file or directory
find: `/work/project/w4m/galaxy4metabolomics/galaxy-dist/database/jobs_directory/000/131/131287/working/FOO': No such file or directory
find: `2': No such file or directory
Warning message:
running command 'find $PWD/FOO 2 -not -name '\.*' -not -path '*conda-env*' -type f -name "*"' had status 1
Error in checkForRemoteErrors(val) :
46 nodes produced errors; first error: invalid UTF-8 input in readChar()
Calls: do.call ... xcmsSet -> xcmsClusterApply -> checkForRemoteErrors
Execution halted
Could not find platform dependent libraries <exec_prefix>
Consider setting $PYTHONHOME to <prefix>[:<exec_prefix>]
Traceback (most recent call last):
File "/work/project/w4m/galaxy4metabolomics/galaxy-dist/database/jobs_directory/000/131/131287/set_metadata_y5adti.py", line 1, in <module>
from galaxy_ext.metadata.set_metadata import set_metadata; set_metadata()
File "/work/project/w4m/galaxy4metabolomics/galaxy-dist/lib/galaxy_ext/metadata/set_metadata.py", line 14, in <module>
import cPickle
ImportError: No module named cPickle
This tag is used to group parameters into sections of the interface. Sections are implemented to replace the commonly used tactic of hiding advanced options behind a conditional, with sections you can easily visually group a related set of options.
https://wiki.galaxyproject.org/Admin/Tools/ToolConfigSyntax#A.3Csection.3E_tag_set
Can we change the tool in order to use RTrange instead of scan range?
It is sometime confusing for users to enter a scan number instead of a RT. users are used to cut their chromatogramme between min and max RT values not between scan numbers. And sometimes scan numbers read in in silicos viewer are not the on read by xcms.
Sometimes during MS files life they are stored under filepaths with accent (e.g in french metabolomics is métabolomique)
and so when converting to mzXML (or other format) sometimes those paths are kept in the file and we get error.
invalid UTF-8 input in readChar() ligne <parentFile fileName="file:///D:/JPA/Laits-2015-06-10-Exactive (Metabolomique R�cap)/./211114031_S5_.raw" for exemple
group function parameters are not complet, the minsamp argument is missing, can we add it in the advanced options list?
Just a warning some deep changes have been added into XCMS and CAMERA for XCMS >1.50, those can affect our tools
With @sneumann we found that is step originally design to delete "é", "è" characters from <parentFile fileName="file://C:/data/métabo/foobar.RAW" where raw files are stored.
2 solutions:
Sometimes there are redundant ion identifiers, especially when using RT in minutes (for example "M123.32T11" for two different ions with mass 123.32 and RT 10.6 min and 11.4 min).
Since identifiers are meant to be unique, something must be done (currently users add more decimal places for mass, change RT in seconds and/or modify identifiers themselves).
I'm currently setting travis test for our tools. I need to run all the tests within 50 minutes and if a test don't produce log alter 10 min, it fails.
https://travis-ci.org/workflow4metabolomics/xcms/builds/124014197
No output has been received in the last 10m0s, this potentially indicates a stalled build or something wrong with the build itself.
The build has been terminated
It seems that the group jobs builded from my test cases are too long for TravisCI.
@yguitton @sneumann or anyone else, do you know which method and/or parameters I can set to reduce executing time?
Or do I have too change my input dataset?
Thanks by advance
The same as for findChromPeaks/xcmsSet parameters.
So far, we don't expose so many arguments for the method Obiwrap.
Thus dear @workflow4metabolomics/ms, (and if you are interesting in: @sneumann, @jotsetung) :
If you want, you can use the same checklist as me (here) and move items.
Many thanks by advanced
Redundancy is created when using dataset collection mode. XCMS summary generates a line of xcmsSet parameters for each datafile (off course they are the same for each file of dataset collection).
xcms_summary_html.zip
The idea is to do the same as decided in #23 for fillpeaks, this time for group module.
Since unzip might not be available on a cluster environment (as in our case on CentOS) it should be in the requirements.
With zip file import the tool failed with a cryptic error:
find: `NA': No such file or directory
find: `/gpfs1/data/galaxy_server/galaxy/jobs_dir/006/6477/working/NA': No such file or directory
Warning message:
running command 'find $PWD/NA -not -name '\.*' -not -path '*conda-env*' -type f -name "*"' had status 1
Error in xcmsSet(NA_character_, nSlaves = 1, method = "matchedFilter", :
No NetCDF/mzXML/mzData/mzML files were found.
Calls: do.call -> do.call -> xcmsSet
Execution halted
The file list returned by unzip seems to be empty and therefore the root directory is undetermined. Checking for this might also be a good idea.
@lecorguille maybe you can include this in your efforts while you are working on the merger anyway
For the "old" version of xcms, since the input param is not named input, the job fails.
My priority before carry on some new developments is to pass the functional tests.
Until recently, the zip format was not enough integrated in Galaxy. But from the 16.01, I have no more excuse 😄
This issue concerns the peak table export in xcms.group and xcms.fillpeaks.
The idea is to:
Indead of the library version
Tested:
Mention them here:
https://github.com/workflow4metabolomics/xcms/blob/dev/galaxy/xcms_xcmsset/abims_xcms_xcmsSet.xml#L71
https://github.com/workflow4metabolomics/xcms/blob/dev/galaxy/xcms_xcmsset/abims_xcms_xcmsSet.xml#L77
https://github.com/workflow4metabolomics/xcms/blob/dev/galaxy/xcms_xcmsset/abims_xcms_xcmsSet.xml#L413
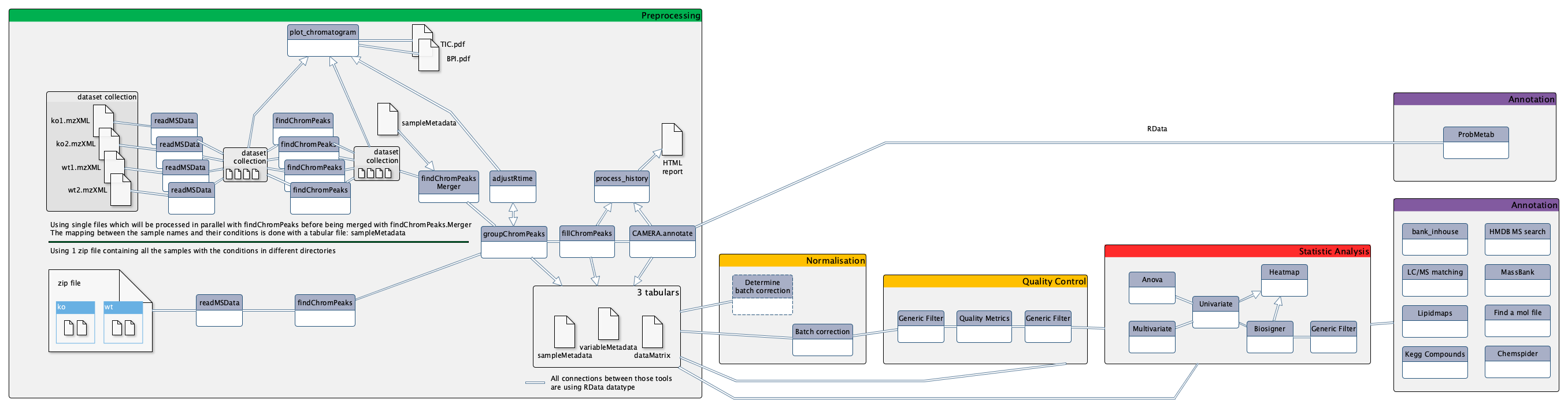
ping @workflow4metabolomics/ms
@lecorguille if we hurry up we might get the datatypes into 17.05 and ready for GCC :)
The unzip datatype can be removed isn't it? This should now be supported by Galaxy naively.
At least for the single file mode
we have a user request regarding xcmsset fitgauss option and verbose.column=TRUE
should be quite easy to add those two options
Note be careful with verbose.column as it will add new columns to xset@peaks tables. those columns can be used only if people do look at each file peak tables individually
Hi I have tried dev version 3.0.0.0 and have a trouble ...
http://galaxydev.workflow4metabolomics.org/u/yguitton/h/xcms30
@lecorguille can you help me
Yann
Hi, tremendous work you're doing! Awesome!
Just wanted to point you to the new Chromatogram/Chromatograms class in MSnbase. It is now very easy to extract ion chromatograms (or base peak or TIC). If you have an OnDiskMSnExp or MSnExp you can simply use the chromatogram method. This returns a Chromatograms object which is simply a matrix like object containing Chromatograms. Rows can be different slices (m/z, rt ranges) of the MS data, columns are for the individual samples/files.
The chromatogram method has also parameters mz and rt that allow to restrict to a certain m/z-rt slice of the MS data. The aggregationFun allows to define how signals for the same rt are handled - for a TIC you would use aggregationFun = "sum", for a BPC aggregationFun = "max".
You can then use the plot method to plot the chromatographic data.
Have also a look at ?MSnbase::chromatogram and ?xcms::chromatogram. For XCMSnExp objects there is an additional parameter adjustedRtime that allows to specify whether the raw or adjusted retention time should be reported.
If a file with a ',' in the file name is used as input then the link is not created properly (the name of the link is the prefix of the filename up to the comma) and xcms does not find input data:
Error in xcmsSet(".", nSlaves = 1, method = "centWave", ppm = 25, peakwidth = c(10, :
No NetCDF/mzXML/mzData/mzML files were found.
Seems to be related to: #65 (some escaping seems to be necessary).
Some exchange with @melpetera
FYI: @chcaron @fgiacomoni
Since xcmsSet (2.1.0) can now accept both a zip file or an individual sample (single file), we can't have the same number of CPU for the 2 type of feeding: \${GALAXY_SLOTS:-1}
There is a system call Dynamic Destination Mapping which will allow to use one or an other <destination> according to some rules (in my case, the value of an argument).
For example, we will use:
I first tried the DTD method but currently, it needs some fix to fit with xcmsSet (PR in progress). It also request the release_16.07 which seem cool but I don't know if my fix will be backported to the 16.07.
I will have to take a look at the Python method.
So W&S
From @ethevenot
J'ai obtenu ce message en lan?ant XCMS :
arguments 'minimized' and 'invisible' are for Windows onlyJ'ai trouvé sur le net l'info suivante :
r-lib/devtools#540
In the xcms.group graphical output (Rplots.pdf), plot names correspond to corresponding mz slices. Mz values are written with 2 decimal places (for exemple "164.94 − 164.96").
The problem is that if you choose for any reason to consider narrow mz slices (changing mzwid to 0.005 for example), then you will have things like "164.95 − 164.95" and will not actually know if it is close or not to your maximum mz width.
Would it be possible for the plot titles to have more decimal places? There is room for longest names, so maybe 4 decimal places would be ok?
So dear @workflow4metabolomics/ms, (and if you have 5 minutes: @sneumann, @jotsetung) :
If you want, you can use the same checklist as me (here) and move items.
Many thanks by advanced
A request we had:
Hi, Is there a way to plot relative intensity on y axis in the BPC and TICs generated from xcmsSet, rather than total intensity?
An example:
sampleMetadata class polarity injOrder sampleType batch
20170209_P_Blanc12 blank positive 38 blank 1
20170209_P_Blanc13 blank positive 52 blank 1
20170209_P_QC01 pool positive 11 pool 1
20170209_P_QC02 pool positive 25 pool 1
20170418_P_S01n01 TV_nd positive 58 sample 1
20170418_P_S01n02 TV_nd positive 69 sample 1
XSET OBJECT INFO
class
20170418_P_Blanc12 <NA>
20170418_P_Blanc13 <NA>
20170418_P_QC01 <NA>
20170418_P_QC02 <NA>
20170418_P_S01n01 TV_nd
20170418_P_S01n02 TV_nd
So the tool should check and raise and error if it met this use-case.
Lack
Wrong
Other
1 - or 2 - in the option labelsDear Santa @lecorguille Claus,
When you use XCMS merger without providing any sampleMetadata file, this means that you have no ready "reference" file for the processing step following xcms analyses.
In addition, it is not always straigthforward to construct the sampleMetadata file, knowing that sample identifiers are raw files' names that can be automatically generated by machine's software with unfriendly automatic names.
The possibility to use an empty sampleMetadata file with already the right identifiers, as it is provided with the zip option, is very handy and reduce significantly the misscase errors compared to manual listing in addition to saving time.
For all this reasons I would strongly recommand to add, when not provided as input, a sampleMetadata file as output with identifiers as first column (of course), and maybe just a second column 'class' with a constant value (for example 'no groups' ou "single group').
Thank you for you time.
M. who behaved really well this year.
ping @yguitton @jfrancoismartin
In the current wrapper version for xcmsSet, scanrange is only available for centWave. In ?xcmsSet, it seems that this obscure option should be available for all methods?
What do you think about that?
Merge/Smach/Squash some intermediate versions within some tools
Crop parts of this big picture to refresh our current images
When running xcms_xcmsset (revision 15646e937936) I get an empty result. The resulting dataset is marked as successful, but its empty.
In the dataset preview I see the following text:
code for methods in class "Rcpp_Ramp" was not checked for suspicious field assignments (recommended package 'codetools' not available?)
I uploaded the input to https://oc.ufz.de/index.php/s/j4aPVY6iwlv6WU7 with password xcms.
Options:
Input: see OC
Scan range option: hide
Extraction method for peaks detection: centWave
Max tolerated ppm m/z deviation in consecutive scans in ppm: 25
Min,Max peak width in seconds: 20,50
Advanced options: hide
Then Galaxy can parallelize the execution.
For backwards compatibility, check at
https://github.com/workflow4metabolomics/xcms/blob/master/src/xcms_w4m_script/xcms.r#L122
if a ZIP or an mzML/netCDF/... is provided.
Then you get one xcmsSet per input file.
After all N xcmsSets are created, use " c(xs1, xs2, xs3, ..." to combine
all individual xcmsSets into one big, like the existing node does.
Yours, Steffen
without have to use CAMERA
So maybe split the tool camera or add a new output within fillpeaks?
Currently 1.44
Available 1.46
We should need to wait for the functionnal tests before go on that #3
I get the following error in xcms.group if I input a sample metadata file in xcms.xcmsSet Merger:
Error in if (!any(gcount >= classnum * minfrac & gcount >= minsamp)) next :
missing value where TRUE/FALSE needed
Calls: do.call ... do.call -> group.density -> group.density -> .local
Any idea what could cause such this problem?
Somehow I have the feeling that this might be related: #59
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.