tanlongzhi / dip-c Goto Github PK
View Code? Open in Web Editor NEWTools to analyze Dip-C (or other 3C/Hi-C) data
Tools to analyze Dip-C (or other 3C/Hi-C) data




























































Hello @tanlongzhi ,
In Dip-C paper, there is a cross section view of the diploid interphase mESC cell 1CDX1-413 and some other cells. The model and its analysis looks good.
In the paper by Nagano et al. (2017) an intercross of Mus musculus 129/SV-Jae and Mus musculus castaneus cells were used. I had a big favor to ask- will it be possible to provide the source for the phased SNPs in VCF file for the mESC diploid cells? and an example pipeline used to generate the 3D models of these diploid mouse cells ? will definitely be great.
I really appreciate your help!
Hi @tanlongzhi ,
Hope you are doing good!
From the notes, what does it mean to rank normalise compartments from 0 to 1?
Will it also be possible to explain how to obtain the plots for fig 4B,E from paper?
From the computed cpg.txt files, dip-c color -c basically interpolates the CG frequencies with the genomic location coordinates in each cell.
I don't understand how to do cell typing based on these obtained .cif files? In the paper you mentioned
Our conclusions held if compartments were defined on the basis of contacts
I might be overthinking it but I suppose you confirmed your PCA clustering based on genomic structure with the plots in fig S17 obtained from contacts?
Any help again will be really great!
Hi Tan,
Happy Monday!
I remember reading in the readme of this repo that hickit is much faster than dip-c. I know that hickit does not implement the 3D imputation step like Dip-c does. But during the read alignment, both use BWA MEM aligner- this aligner is based on BWT/FM-index and gives improved performance but a large index build up time.
I ask these questions because instead of working on a single cell, I wonder how much time it will take to generate many models from many cells. Generating m number of models will help in downstream analysis.
I know that we can generate m models for a given cell explicitly #5 (comment) , and we could do this in parallel for many cells?
Thanks again!
Hi Longzhi,
I am very interested in learning construct 3D model from HiC data. After a long search, I recently find your fantastic work. I have downloaded fastq file for for GM12878 cell 1 from https://www.ncbi.nlm.nih.gov/sra/SRX4133191 , and now try to follow the instruction in this repo. However, I have problems during the further imputing steps:
"con_to_ncc.sh impute.con.gz
nuc_dynamics.sh impute.ncc 0.1
dip-c impute3 -3 impute.3dg clean.con.gz | gzip -c > impute3.round1.con.gz
dip-c clean3 -c impute.con.gz impute.3dg > impute.clean.3dg
con_to_ncc.sh impute3.round1.con.gz
nuc_dynamics.sh impute3.round1.ncc 0.1
dip-c impute3 -3 impute3.round1.3dg clean.con.gz | gzip -c > impute3.round2.con.gz
dip-c clean3 -c impute3.round1.con.gz impute3.round1.3dg > impute3.round1.clean.3dg
con_to_ncc.sh impute3.round2.con.gz
nuc_dynamics.sh impute3.round2.ncc 0.1
dip-c impute3 -3 impute3.round2.3dg clean.con.gz | gzip -c > impute3.round3.con.gz
dip-c clean3 -c impute3.round2.con.gz impute3.round2.3dg > impute3.round2.clean.3dg
...
"
I feel like the cleaned 3dg file by clean3 at each step is not involved in the next round. The reason I start to question about this is actually because one error I encountered:
dip-c impute3 -3 GM12878_cell1_dipc_phased.clean.impute.clean.3dg GM12878_cell1_dipc_phased.clean.con.gz | gzip -c > GM12878_cell1_dipc_phased.impute3.round1.con.gz
[M::impute3] read a 3D structure with 55404 particles at 100000 bp resolution
[M::impute3] read 612536 contacts (82.47% intra-chromosomal, 8.94% legs phased)
[M::classes] imputed haplotypes for chromosome pair (13,17): 392 contacts (85.2% phased)
[M::classes] imputed haplotypes for chromosome pair (5,8): 1679 contacts (97.74% phased)
[M::classes] imputed haplotypes for chromosome pair (16,17): 216 contacts (66.2% phased)
[M::classes] imputed haplotypes for chromosome pair (1,20): 1078 contacts (92.76% phased)
Traceback (most recent call last):
File "dip-c", line 130, in
main()
File "dip-c", line 63, in main
return_value = impute3.impute3(sys.argv[1:])
File "impute3.py", line 109, in impute3
con_data.impute_from_g3d_data(g3d_data, max_impute3_distance, max_impute3_ratio, max_impute3_ratio * g3d_resolution, is_male, par_data, vio_file)
File "classes.py", line 907, in impute_from_g3d_data
self.con_lists[ref_name_tuple].impute_from_g3d_data(g3d_data, max_impute3_distance, max_impute3_ratio, min_impute3_separation, is_male, par_data, vio_file)
File "classes.py", line 757, in impute_from_g3d_data
con.impute_from_g3d_data(g3d_data, max_impute3_distance, max_impute3_ratio, min_impute3_separation, is_male, par_data, vio_file)
File "classes.py", line 544, in impute_from_g3d_data
impute3_ratio = impute3_distance / con_distance_tuples[1][1]
TypeError: unsupported operand type(s) for /: 'NoneType' and 'NoneType'
Here are the head lines from two input files:
head GM12878_cell1_dipc_phased.clean.impute.clean.3dg
1(mat) 1200000 7.95772097608 -12.0072914165 6.67592442321
1(mat) 1300000 8.89210987528 -11.4486456224 6.61131843187
1(mat) 1400000 8.8277193141 -10.3798272863 6.83290065793
1(mat) 1500000 8.10570766598 -9.67144265436 6.35097905003
1(mat) 1600000 7.99275487247 -8.53433974384 6.52683266786
1(mat) 1700000 6.70429668241 -8.61794012705 5.86833067325
1(mat) 1800000 5.62631622929 -8.49098630055 5.17833888961
1(mat) 1900000 4.8879961287 -8.44522282731 3.98121528589
1(mat) 2000000 3.80732676666 -7.76977419875 3.35459947567
1(mat) 2100000 3.06260319638 -8.19929825445 4.23444940641
zcat GM12878_cell1_dipc_phased.clean.con.gz | head
1,756415,. 1,1095231,.
1,757502,. 1,1218674,.
1,815689,. 1,1186165,.
1,818341,. 1,862101,.
1,830604,. 1,835996,.
1,839037,. 1,858631,.
1,848406,. 1,850417,.
1,858704,. 1,861316,.
1,861508,. 1,862932,.
1,918117,1 1,1231475,.
Here are the command I used to construct the input files:
seqtk mergepe SRR7226683_1.fastq SRR7226683_2.fastq | lianti trim - | bwa mem -Cp bwa_index_rmchr/Homo_sapiens_assembly19.fasta - | samtools view -uS | sambamba sort -o GM12878_cell1_dipc_rmchr.bam /dev/stdin
dip-c seg -v snps/NA12878.txt.gz GM12878_cell1_dipc_rmchr.bam | gzip -c > GM12878_cell1_dipc_phased.seg.gz
dip-c con GM12878_cell1_dipc_phased.seg.gz | gzip -c > GM12878_cell1_dipc_phased.con.gz
dip-c dedup GM12878_cell1_dipc_phased.con.gz | gzip -c > GM12878_cell1_dipc_phased.dedup.gz
dip-c reg -p hf GM12878_cell1_dipc_phased.dedup.gz | gzip -c > GM12878_cell1_dipc_phased.reg.con.gz
dip-c clean GM12878_cell1_dipc_phased.dedup.gz | gzip -c > GM12878_cell1_dipc_phased.clean.con.gz
dip-c impute GM12878_cell1_dipc_phased.clean.con.gz | gzip -c > GM12878_cell1_dipc_phased.clean.impute.con.gz
con_to_ncc.sh GM12878_cell1_dipc_phased.clean.impute.con.gz
nuc_dynamics.sh GM12878_cell1_dipc_phased.clean.impute.ncc 0.1
Thanks a lot!
Looking forward to your help!
Bo Zhang
Hi Tan,
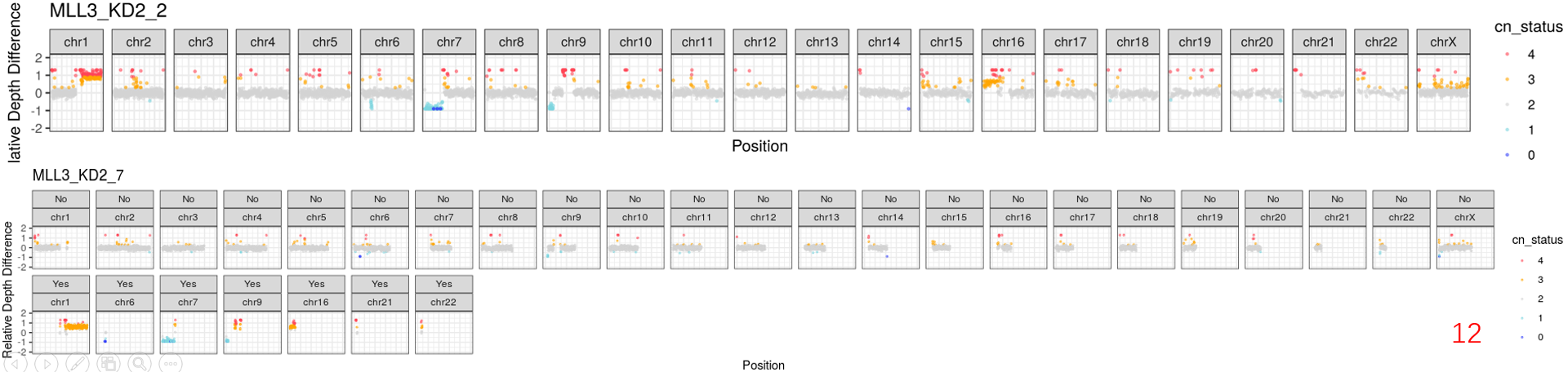
it's really a interesting tool to analyse the 3D genome on single-cell level. Recently, we also implemented this platform in our projects. But we met some problems. As you can see in some cells of our data, there were obvious copy-number (CN) gains or losses regions. Although you mentioned .reg files in README.md file, I'm not sure where and how should we do to remove them.
Now, I have two plans to achive it: 1) remove the CN regions by samtools and bedtools from *.aln.sam.gz files; 2) remove them by dip-c reg3. But I'm not sure which way is better, because the RMSD is the important standard for the structure. And the RMSD scores would be calculated based on *3dg files. So, I prefer to remove these region by the first way to before genome structure reconstruction. Do you think whether it's resonbale to do?
Sincerely
Xiangyu Pan
West China hospital
Sichuan University
Chengdu, China
I ran the tutorial hickit -s${rep} -M -i ... et cetera and got the following error: hickit: invalid option -- 'M'
Looks like -M is no longer an option for hickit. Do you know what its purpose was in this command? Thanks!
Hi,
I want to perform Dip-C for home-made cell line. We don't have the SNP information in this cell line. So, I have two questions:
Best wishes
Hanwen
Sorry for going back and forth between theories but I had some general questions:
The statistical property for inter-chromosomal/ long range intra-chromosomal contacts- explained is definitely convincing, but can we show the same via the plot between contact probability Vs genomic distance? I know that for intra-chromosomal- we should expect a slope of -1(fractal globule case), for inter-chromosomal- should be expect a slope of -2? How to find the contact probability in this case?
Coming back to #4 (comment) , you remove short range contacts with unknown haplotypes on both legs. Won't the either or both strategy help in producing more better 3D structures?
In Dip-C, Imputation in done in 2D and then 3D. IN the supplementary material you mentioned
Imputation would occur if the winning haplotype tuple (the shortest 3D distance) yielded a 3D distance ≤ 20 particle radii and ≤ 0.5 times the second shortest 3D distance.
I am a bit confused with the second shortest 3D distance? Could you explain the 3D imputation in general?
--> Thinking along the same lines, are there any limitations in the current methods you think might be improved on? In the Deep learning age, can it help current 3D/4D genome modeling and analysis even further?
Thanks again, I really appreciate it!
Hi Tan, @tanlongzhi
When i try to colored the chromosomes and cpg island, the command run successfully, but can not write the colors.
Can you help me to solve this problem?
Commond:
dip-c color -c $dip_c/color/mm10.cpg.20k.txt patski.G2M.3dg
patski.G2M.3dg:

mm10.cpg.20k.txt:

Results:

Hi Tan,
This's Yuyang from Tsinghua Uni, Beijing.
Hope you have a nice holiday.
I just followed the "Typical Workflow" in my server and got trouble at the very beginning step.
../seqtk-master/seqtk mergepe SRR7226685_1.fastq SRR7226685_2.fastq | ../lianti-master/lianti trim - |../bwa-master/bwa mem -Cp ../hg19.fa - | samtools view -uS |../sambamba-0.6.8-linux-static sort -o aln.bam /dev/stdin
./dip-c seg -v snps/NA12878.txt.gz aln.bam | gzip -c > phased.seg.gz
It throw an error in the second step.
[M::seg] pass 2: read 24000000 alignments, last at chrX:33183726
[M::seg] pass 2: read 24100000 alignments, last at chrX:45099699
[M::seg] pass 2: read 25000000 alignments, last at chrX:149610521
[M::seg] pass 2: read 25100000 alignments, last at chrY:13801064
[M::seg] pass 2: read 25200000 alignments, last at chr9_gl000198_random:71282
[M::seg] pass 2: read 25300000 alignments, last at chrUn_gl000216:27250
[M::seg] pass 2: read 25400000 alignments, last at chrUn_gl000220:139949
[M::seg] pass 2: read 25500000 alignments, last at chrUn_gl000226:14258
[M::seg] pass 2: read 25600000 alignments, last at *
[M::seg] pass 2: read 26800000 alignments, last at *
[M::seg] pass 2: read 26900000 alignments, last at *
[M::seg] pass 2: read 27000000 alignments, last at *
[M::seg] pass 2: cleaning 2230534 candidate reads
[M::seg] pass 2 done: read 27005726 alignments; kept 1815407 candidate reads (6.72% of alignments)
Traceback (most recent call last):
File "./dip-c", line 130, in
main()
File "./dip-c", line 42, in main
return_value = seg.seg(sys.argv[1:])
File "/home/DAILY_WORK/LYY/dip-c-master/seg.py", line 129, in seg
for pileup_column in bam_file.pileup(snp_chr, snp_locus - 1, snp_locus):
File "pysam/libcalignmentfile.pyx", line 1314, in pysam.libcalignmentfile.AlignmentFile.pileup (pysam/libcalignmentfile.c:16452)
File "pysam/libchtslib.pyx", line 675, in pysam.libchtslib.HTSFile.parse_region (pysam/libchtslib.c:11863)
ValueError: invalid contig `1
By the way, could you mind uploading some key intermediate files? It would make the pipeline easy to follow and also for debugging. For example, in the "Interactive Visualization of 3D Genomes" section, cell.3dg is used in the whole section to make the pretty figures. Moreover, the 3D reconstruction process seems a little bit tricky as you also showed in the Fig. S8 in your Science paper. Do you have any suggestions to gain a reasonable simulated 3D structure?
Thanks so much for your help!
Yuyang
Hi @tanlongzhi ,
In exp,py, will it possible to include an option for translating the chromosomes instead of the two homologs separately? I mean instead of finding the centers of each homolog, can we find the centers of the homolog pair and then translate.
Hi Tan,
The models look great and a single model is good for visualization purposes. I remember that in the paper you did analysis on some more replicates to get the results. And nuc_dynamics has -m options to define the number of models we wish to find. Thinking along the same lines, can we specify the number of independent models to be generated using dip-c/ hickit?? Say for each diploid single cells I want 100 models so as to do analysis on structural positioning or pairwise distance distributions between loci??
Hi @tanlongzhi ,
I was also trying to visualize the mouse diploid genome using the output _.impute3.round4.clean.3dg files from the GEO database for 1CDX1-413 diploid cell by using the following command:
(py2) Taraks-MacBook-Pro:dip-c tarakshisode$ ./dip-c color -n ./NXT-43.250kb/mm9.chr.txt ./CDX1-413.20k/CDX1-413.20k.dip-c.3dg | dip-c vis -c /dev/stdin ./CDX1-413.20k/CDX1-413.20k.dip-c.3dg > ./CDX1-413.20k/CDX1-413.20k.dip-c.cif
Note that I am using mm9.chr.txt with chr1 through chrX
I get the following error:
.
.
[M::color] analyzed 236000 particles (97.85%)
[M::color] analyzed 237000 particles (98.27%)
[M::color] analyzed 238000 particles (98.68%)
[M::color] analyzed 239000 particles (99.1%)
[M::color] analyzed 240000 particles (99.51%)
[M::color] analyzed 241000 particles (99.92%)
[M::color] writing 241181 colors (100.0%)
Traceback (most recent call last):
File "./dip-c", line 122, in <module>
main()
File "./dip-c", line 73, in main
return_value = color.color(sys.argv[1:])
File "/Users/tarakshisode/dip-c/color.py", line 246, in color
sys.stdout.write("\t".join([hom_name, str(ref_locus), str(color_data[(hom_name, ref_locus)])]) + "\n")
IOError: [Errno 32] Broken pipe
Any help will be great!
Hi @tanlongzhi!
I'm attempting to work with the final 3D genome txt files from the 2019 publication, downloaded from (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE121791), and the GEO site says 'please see 00README.md' to find information on data processing. In the similar GEO site for the 2018 publication, the 'data processing' section mentions that the final 3D genome files for each cell are called 'impute3.round4.3dg', for example, 'GSM3271347_gm12878_01.impute3.round4.3dg.txt'. However, the files in the 2019 publication do not have the same format. I see files such as 'GSM3446115_cell_014.20k.2.clean.3dg.txt', are these the final 3D genome files? Where can I find this 00README.md, assuming that it is different from the README.md and README_old.md in this repo?
Thank you so much!
Hey @tanlongzhi,
Is there a way to highlight the centromere positions in the movie of the 3D models for human/mouse? I ask that because it may help us determine the cell cycle phase.
Hi Tan,
Dip-c indeed has a lot of relevant analysis which are really useful for me. I was trying out to produce results similar to the ones you have in fig 4A,C in your paper. There is a lot to learn from these figures and also how to generate them(like cross section plots for centromere to telomere organisation, intermingling plots)
I was reading up on it in supplementary file but could not follow clearly. It would be great if there is a possibility to provide a sample algorithm for generating these figures?
Thanks again!
Hey @tanlongzhi ,
Hope you are doing good!
In your analysis of interchromosomal and long-range intrachromosomal contacts, how did you pileup the inter chromosomal contact maps to generate the super elliptical neighbourhood region? Because the dimensions for each chromosomes are different, I don't know how to overlay the haplotype resolved inter chromosomal contact maps?
Any help will be great!
Hi @tanlongzhi
Thanks for your powerful Dip-c tools. I am looking for a way to generate a 3D proximity map like the one you mentioned in your article.
A 3D proximity map—a matrix of fractions of cells where each pair of 20-kb particles are nearby in 3D (distance ≤ 3.0 radiiwas then generated by binarizing each single-cell matrix of pairwise 3D distances and averaging them, with “scripts/threshold_np_float.py” of the “dip-c” package (“scripts/threshold_np_float.py 3”).
And I did not find such scripts in the files. I guess it may be moved to somewhere else. Could you please tell me where it is? Thank you.
Also, I am trying to figure out the TADs in single cells and I saw TAD.py in this file. Is the output file of dip-c tad-l shows the exact region of TAD regions? And is there a way to visualize such a TAD result file? Any information or guidance is really appreciated. Thank you.
Yuchen
Thanks for providing this awesome tool!
I just want to double-check do I need to use variant masked genome ref when doing the alignment? That's what I read in some allele-specific analysis paper.
Sorry, I do not fully understand how hickit.js resolve the phrase.
Thanks!
Hey @tanlongzhi ,
I didn't understand the role of the following line of code in the modified nuc_dynamics.py file
digits_upper = "0123456789ABCDEFGHIJKLMNOPQRSTUVWXYZ"
digits_lower = digits_upper.lower()
digits_upper_values = dict([pair for pair in zip(digits_upper, range(36))])
digits_lower_values = dict([pair for pair in zip(digits_lower, range(36))])
def encode_pure(digits, value):
"encodes value using the given digits"
assert value >= 0
if (value == 0): return digits[0]
n = len(digits)
result = []
while (value != 0):
rest = value // n
result.append(digits[value - rest * n])
value = rest
result.reverse()
return "".join(result)
def decode_pure(digits_values, s):
"decodes the string s using the digit, value associations for each character"
result = 0
n = len(digits_values)
for c in s:
result *= n
result += digits_values[c]
return result
def hy36encode(width, value):
"encodes value as base-10/upper-case base-36/lower-case base-36 hybrid"
i = value
if (i >= 1-10**(width-1)):
if (i < 10**width):
return ("%%%dd" % width) % i
i -= 10**width
if (i < 26*36**(width-1)):
i += 10*36**(width-1)
return encode_pure(digits_upper, i)
i -= 26*36**(width-1)
if (i < 26*36**(width-1)):
i += 10*36**(width-1)
return encode_pure(digits_lower, i)
raise ValueError("value out of range.")
def hy36decode(width, s):
"decodes base-10/upper-case base-36/lower-case base-36 hybrid"
if (len(s) == width):
f = s[0]
if (f == "-" or f == " " or f.isdigit()):
try: return int(s)
except ValueError: pass
if (s == " "*width): return 0
elif (f in digits_upper_values):
try: return decode_pure(
digits_values=digits_upper_values, s=s) - 10*36**(width-1) + 10**width
except KeyError: pass
elif (f in digits_lower_values):
try: return decode_pure(
digits_values=digits_lower_values, s=s) + 16*36**(width-1) + 10**width
except KeyError: pass
raise ValueError("invalid number literal.")
Any help will be great!
Hi,tan
If I don't have the SNP information file, can I use the dip-c process?
Thanks
Hey @tanlongzhi ,
How would one use the align.py script? What all inputs can we give and what outputs can we get?
Is it possible to get a genome wide RMSD score?
Thanks!
Hi Longzhi,
Is the NA12878.txt.gz under snps folder the correct file for imputing Gm12878 single cell hic? Or it is just an example file? If it is the case, do you mind share the full SNP information?
Thanks
Bo
Hi @tanlongzhi
Thanks for your powerful hickit and dip-c package. It is great. I met the same problem as issue3 when I tried to convert my .3dg file to a cif file using this code
dip-c color -n color/hg19.chr.txt cell.3dg | dip-c vis -c /dev/stdin cell.3dg > cell.n.cif
And get an error like
python ./dip-c/dip-c color -n hg19.chr.txt clear_pbmc.3dg
Traceback (most recent call last):
File ".../dip-c", line 130, in <module>
main()
File ".../dip-c/dip-c", line 75, in main
return_value = color.color(sys.argv[1:])
File ".../dip-c/color.py", line 218, in color
g3d_data = file_to_g3d_data(open(args[0], "rb"))
File ".../dip-c/classes.py", line 1427, in file_to_g3d_data
g3d_data.add_g3d_particle(string_to_g3d_particle(g3d_file_line.strip()))
File ".../dip-c/classes.py", line 1214, in string_to_g3d_particle
hom_name, ref_locus, x, y, z = g3d_particle_string.split("\t")
TypeError: a bytes-like object is required, not 'str'
I have already used the grep -v "^#" hickit.3dg ' > dip-c.3dg code to delete the headers and I am using diploid data.
I try to use NXT-43.250kb.hickit.3dg.zip from tarak77 to figure out the problem. After using his file and grep -v "^#" hickit.3dg | awk -F'[\t,]' -v OFS='\t' '{print $1 "(pat)",$2,$3,$4,$5}' > dip-c.3dg. I still get the same error.
I also download the Dip-c original dataset from GSM3271347 and process the "GSM3271347_gm12878_01.impute3.round4.clean.3dg.txt.gz" file to figure out the issue and the problem still exists when I want to convert this file to a cif file.
python ./dip-c/dip-c color -n hg19.chr.txt impute3.round4.clean.3dg.txt
Traceback (most recent call last):
File "../dip-c/dip-c", line 130, in <module>
main()
File ".../dip-c/dip-c", line 75, in main
return_value = color.color(sys.argv[1:])
File ".../dip-c/color.py", line 218, in color
g3d_data = file_to_g3d_data(open(args[0], "rb"))
File ".../dip-c/classes.py", line 1427, in file_to_g3d_data
g3d_data.add_g3d_particle(string_to_g3d_particle(g3d_file_line.strip()))
File ".../dip-c/classes.py", line 1214, in string_to_g3d_particle
hom_name, ref_locus, x, y, z = g3d_particle_string.split("\t")
TypeError: a bytes-like object is required, not 'str'
Could you please help me with that? Any kind of help is really appreciated.
Wish you a great day.
Hi @tanlongzhi ,
I just want to make the pipeline work in my server, so I tested following code with the file you provided (tab-delimited).
dip-c color -c color/hg19.cpg.20k.txt cell.3dg | dip-c vis -M -c /dev/stdin cell.3dg > cell.cpg.cif
However, it threw an error like:
[M::color] read a 3D structure with 10 particles at 20000 bp resolution
[M::vis] read a 3D structure with 10 particles at 20000 bp resolution
Traceback (most recent call last):
File "./dip-c", line 130, in
main()
File "./dip-c", line 75, in main return_value = color.color(sys.argv[1:])
File "/home/XShenLab/DAILY_WORK/LYY/dip-c-master/color.py", line 186, in color
color = ref_name_ref_locus_colors[(g3d_particle.get_ref_name(), g3d_particle.get_ref_locus())]
File "/home/XShenLab/DAILY_WORK/LYY/dip-c-master/classes.py", line 1181, in get_ref_name
return hom_name_to_ref_name_haplotype(self.hom_name)[0]
File "/home/XShenLab/DAILY_WORK/LYY/dip-c-master/classes.py", line 40, in hom_name_to_ref_name_haplotype
ref_name, haplotype = hom_name.split("(") ValueError: need more than 1 value to unpack
[M::main] command: ./dip-c vis -M -c /dev/stdin cell.3dg
[M::main] finished in 0.8 sec
cell.3dg:
1 1420000 0.791377837 10.99472914 -13.18828977
1 1440000 -0.268241284 10.52008759 -13.08962573
1 1460000 -1.385307524 10.55137875 -13.14401422
1 1480000 -1.559841017 11.43408291 -13.60263012
1 1500000 -0.770991778 11.47584885 -14.58811372
1 1520000 -0.084824511 12.26246908 -14.35428963
1 1540000 -0.458643807 12.59857918 -13.47011493
1 1560000 -0.810322906 12.2461644 -12.31729334
1 1580000 -2.08211172 12.88868387 -12.87420078
1 1600000 -3.520939482 13.18509354 -12.41186844
Thanks for your help.
Hi Tan,
This may seem a random question, but I have seen in many papers before where there are movies of chromosome expanding/exploding from the genome structures just to make the visualization more appealing. I noticed that you too have the expand/explode figure 2A in your paper. Could you share how to generated those models, i.e. is there a way to expand/explode the genome model so as to clearly see each of the chromosomal pairs ?
Hi Tan @tanlongzhi , I downloaded some .3dg file from your GEO repository, i found most of them are based on 20kb resolution.
Is it possible I can convert them to 40kb, 80kb without remodeling? i was thinking if convert to 40kb for example, we can use the center point of 2 points (atoms) from 20kb file? does this make sense?
Hi Longzhi,
I want to apply Hg38 version genome to analyze Dip-C data.
But I cannot find the suitable cpg.1m.txt file or cpg.20k.txt files for Hg38.
Could you teach me how to generate these files?
Hanwen
2022/05/27
Hi Longzhi,
I used the latest version of hickit to perform the single-cell Hi-C data, when I extracted segments with the following cold
k8 hickit.js sam2seg -v $snp ${sp}_aln.sam.gz | hickit.js chronly -y - | gzip > ${sp}_contacts_seg.gz
I got the following data. It's strange that the read format is not like yours provided in dip-c README.
ST-E00522:513:HVKWHCCXY:5:1101:15676:1854 chr3!119719306!119719456!-!.!60!1 chr3!119713160!119713310!+!.!60!1 ST-E00522:513:HVKWHCCXY:5:1101:18933:1872 chr18!8795309!8795364!-!.!34!1 chr18!9160818!9160929!+!.!60!2 ST-E00522:513:HVKWHCCXY:5:1101:6877:2083 chr19!26243774!26243961!-!.!60!2 chr19!26197893!26197970!+!.!60!1
Hello,
It is a perfect timing to get hold of a pipeline which will help us produce 3D structures of single diploid cells. Thank you for that!
I have worked with the methods in literature for producing good 3D structures for my mESCs data for haploid interphase cells- Chrom3D, SIMBA3D, ISDHIC and Tim Stevens nuc_process/nuc_dynamics pipeline (which starts with FASTQ files). Tim Stevens procedure could also be used on diploid cells but it will not generate all the 40 chromosomes as the diploid contact maps cannot be disentangled into maternal and paternal maps, yet.
Recently I came across your paper Dip-C and the results showed just what I wanted.
I am a bit unclear about the workflow described for generating the diploid 3D structures. Is it possible adapt it to the nuc_dynamics pipeline? Any help will be great!
Thanks,
Tarak
Hi Tan,
I was going over haplotype imputation step(2D) from the supplementary material and was a bit unclear with the figure S7E. Missing haplotype of a given contact could be imputed from haplotypes of other contacts in its superelliptical neighborhood. And this is based on a voting system. What exactly does n in the same figure represent? And why for n=90 it is uninformative?
Again, intrachromosomal contacts that had unknown haplotypes on both legs were not imputed but inter chromosomal were?
I want to remove repetitive (contact-less) regions, so I run the following command:
for rep in seq 1 3
do
/home/lihaoxing/dip-c-master/scripts/hickit_3dg_to_3dg_rescale_unit.sh 20k.${rep}.3dg
/home/lihaoxing/dip-c-master/dip-c clean3 -c 4-262-1.impute.con.gz 20k.${rep}.dip-c.3dg > 20k.${rep}.clean.3dg # remove repetitive (contact-less) regions
done
and then there is a problem, how should I solve it?
Traceback (most recent call last):
File "/home/lihaoxing/dip-c-master/dip-c", line 130, in
main()
File "/home/lihaoxing/dip-c-master/dip-c", line 65, in main
import clean3
File "/home/lihaoxing/dip-c-master/clean3.py", line 5, in
from classes import Haplotypes, LegData, ConData, file_to_con_data, Leg, Par, ParData, G3dData, file_to_g3d_data
File "/home/lihaoxing/dip-c-master/classes.py", line 6, in
from scipy import interpolate
File "/home/lihaoxing/miniconda3/lib/python3.9/site-packages/scipy/init.py", line 131
raise ImportError(msg) from e
^
SyntaxError: invalid syntax
Hi @tanlongzhi ,
Previously I asked you about how you obtained the pileup of inter chromosomal contacts to see the ellipsoidal shape, which you then use for imputation. I know you told to use ard.py script for doing so. Would it be possible to explain what the script is doing in general? How are you defining a reference contact and how are you piling them up?
Thanks again!
Hi Longzhi,
I sorry to bother you again.
Our approach to recording a base in the genome can be divided into two group: 0-base or 1-base.
I would like to know the coordinate system used by the SNP file you provided in Github.
Best wishes~
Hanwen
Hi Tan,
I was trying to visualize one of the diploid cell contacts with and without haplotype imputation using the Juicebox tool. The haplotype-resolved .con file gives a visualization but the without haplotype resolved .con file gives me the following error:
wg-dhcp174d163d001:fraser-lab tarakshisode$ java -Xmx2g -jar juicer_tools.1.7.6_jcuda.0.8.jar pre -n ../dip-c/CDX1-413.20k/GSE117109_GSM2476407_1CDX1-413.dedup.juicer.txt.gz ../dip-c/CDX1-413.20k/1CDX1-413.dedup.hic mm9
Not including fragment map
Start preprocess
Writing header
Writing body
java.lang.RuntimeException: No reads in Hi-C contact matrices. This could be because the MAPQ filter is set too high (-q) or because all reads map to the same fragment.
at juicebox.tools.utils.original.Preprocessor$MatrixZoomDataPP.mergeAndWriteBlocks(Preprocessor.java:1472)
at juicebox.tools.utils.original.Preprocessor$MatrixZoomDataPP.access$000(Preprocessor.java:1243)
at juicebox.tools.utils.original.Preprocessor.writeMatrix(Preprocessor.java:656)
at juicebox.tools.utils.original.Preprocessor.writeBody(Preprocessor.java:378)
at juicebox.tools.utils.original.Preprocessor.preprocess(Preprocessor.java:286)
at juicebox.tools.clt.old.PreProcessing.run(PreProcessing.java:105)
at juicebox.tools.HiCTools.main(HiCTools.java:98)
Am I doing something wrong?
I have attached the deduce.juicer.txt.gz file, just in case
GSE117109_GSM2476407_1CDX1-413.dedup.juicer.txt.gz
Thanks again for the contact matrix section!
contact.pairs or impute.pairs files(from hickit repo) to the required input .con files required for bincon .con files(for haploid cell data) and still generate these contact matrices?Hi Longzhi,
I'm sorry to bother you again.
We adopted the library contruction method in "SCIENCE" article to generate our Dip-C data.
But, The pipeline you provided run successfully, but I met some problems.
For now, I doubt our library contruction may not be rightful.
But, I don't know how to perform quality control on our data, except sequencing quality.
Honestly, we should use the same cell line in "SCIENCE" article to check if the library contruction was successful.
But I'm not the man to do it.
Can you give me some suggestions?
Hanwen
Best wishes ~
Hi Longzhi,
I really appreciate all the help you provide. I really like the 3D model you build in the original science paper. Now the biggest question in my mind is can I apply dip-c software on normal population hic? If not, what would be the key hurdle for preventing that happen?
Thanks
Bo
Hey @tanlongzhi ,
Will it be possible to pass in particle size list with resolutions higher than 20kb? what would cause it to fail?
Thanks!
Hey @tanlongzhi ,
This might be similar to issue #26 (comment) but i was trying to model the data from an unfertilized egg cell.
To do so, I use hickit for the initial preprocessing steps and then for 3D reconstruction and imputation I use this repo.
To use this repo I convert the obtained impute.pairs and contact.pairs files from hickit to .con files by
using hickit_pairs_to_con.sh and hickit_impute_pairs_to_con.sh scripts.
During the first round of 3D reconstruction, I am seeing that the .3dg file has both (mat) and (pat) rows. I find this strange because the .ncc file only has one haplotype notation (pat).
Also note here that even though the data is from an egg cell, my 0 is actually mat and 1 is pat. Because I messed up while defining the SNP file, so thats why i have (pat) in my .ncc file.
So thats why maybe when implementing the dip-c impute3 -3 code, i get an error
Traceback (most recent call last):
File "/home/tshisode/dip-c/dip-c", line 130, in <module>
main()
File "/home/tshisode/dip-c/dip-c", line 63, in main
return_value = impute3.impute3(sys.argv[1:])
File "/home/tshisode/dip-c/impute3.py", line 109, in impute3
con_data.impute_from_g3d_data(g3d_data, max_impute3_distance, max_impute3_ratio, max_impute3_ratio * g3d_resolution, is_male, par_data, vio_file)
File "/home/tshisode/dip-c/classes.py", line 907, in impute_from_g3d_data
self.con_lists[ref_name_tuple].impute_from_g3d_data(g3d_data, max_impute3_distance, max_impute3_ratio, min_impute3_separation, is_male, par_data, vio_file)
File "/home/tshisode/dip-c/classes.py", line 757, in impute_from_g3d_data
con.impute_from_g3d_data(g3d_data, max_impute3_distance, max_impute3_ratio, min_impute3_separation, is_male, par_data, vio_file)
File "/home/tshisode/dip-c/classes.py", line 544, in impute_from_g3d_data
impute3_ratio = impute3_distance / con_distance_tuples[1][1]
TypeError: unsupported operand type(s) for /: 'NoneType' and 'float'
Again, your help will be great!
Below i have attched the files i mentioned above.
Hi Tan,
Initially I was using hickit for my mESC diploid cell data and got results but the 3D models were a bit non contiguous, since I was using a high resolution for less contacts (lh3/hickit#5 (comment)) , but now was trying to use dip-c for the same data.
Since I don't have META reads, I skip the lianti trim - option in the command below and get an error:
(py2) wg-dhcp174d163d001:seqtk tarakshisode$ ./seqtk mergepe ../nuc_processing-master/data/lane4559_AGGCAGAA_CTCTCTAT_1CDX1-25_L001_R1.fq.gz ../nuc_processing-master/data/lane4559_AGGCAGAA_CTCTCTAT_1CDX1-25_L001_R4.fq.gz | bwa mem -Cp ../GRCm38.p6.genome.fa - | samtools view -uS | sambamba sort -o ../dip-c/1CDX1-25.new/aln.bam /dev/stdin
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 66226 sequences (10000126 bp)...
[M::process] 0 single-end sequences; 66226 paired-end sequences
[M::process] read 66226 sequences (10000126 bp)...
[M::mem_pestat] # candidate unique pairs for (FF, FR, RF, RR): (474, 15653, 432, 453)
[M::mem_pestat] analyzing insert size distribution for orientation FF...
[M::mem_pestat] (25, 50, 75) percentile: (1403, 3030, 5782)
[M::mem_pestat] low and high boundaries for computing mean and std.dev: (1, 14540)
[M::mem_pestat] mean and std.dev: (3759.79, 2708.84)
[M::mem_pestat] low and high boundaries for proper pairs: (1, 18919)
[M::mem_pestat] analyzing insert size distribution for orientation FR...
[M::mem_pestat] (25, 50, 75) percentile: (173, 241, 328)
[M::mem_pestat] low and high boundaries for computing mean and std.dev: (1, 638)
[M::mem_pestat] mean and std.dev: (250.96, 110.76)
[M::mem_pestat] low and high boundaries for proper pairs: (1, 793)
[M::mem_pestat] analyzing insert size distribution for orientation RF...
[M::mem_pestat] (25, 50, 75) percentile: (1496, 3001, 5646)
[M::mem_pestat] low and high boundaries for computing mean and std.dev: (1, 13946)
[M::mem_pestat] mean and std.dev: (3699.66, 2695.92)
[M::mem_pestat] low and high boundaries for proper pairs: (1, 18096)
[M::mem_pestat] analyzing insert size distribution for orientation RR...
[M::mem_pestat] (25, 50, 75) percentile: (1581, 3400, 5536)
[M::mem_pestat] low and high boundaries for computing mean and std.dev: (1, 13446)
[M::mem_pestat] mean and std.dev: (3817.76, 2657.10)
[M::mem_pestat] low and high boundaries for proper pairs: (1, 17401)
[M::mem_pestat] skip orientation FF
[M::mem_pestat] skip orientation RF
[M::mem_pestat] skip orientation RR
[M::mem_process_seqs] Processed 66226 reads in 75.280 CPU sec, 75.835 real sec
[M::process] 0 single-end sequences; 66226 paired-end sequences
[E::sam_parse1] unrecognized type :
[W::sam_read1] Parse error at line 141
[main_samview] truncated file.
But since I had a aln.sam file from hickit, I convert it to aln.bam and use the command for identifying genomic contacts which again leads to an error:
(py2) wg-dhcp174d163d001:dip-c tarakshisode$ ./dip-c seg -v ../hickit/cast_129.phased_SNP.tsv.gz ./1CDX1-25.new/aln.bam | gzip -c > ./1CDX1-25.new/phased.seg.gz
[M::seg] pass 1: read 100000 alignments, last at chr1:29284440; added 16587 candidate reads
[M::seg] pass 1: read 200000 alignments, last at chr9:9941957; added 33426 candidate reads
[M::seg] pass 1: read 300000 alignments, last at chr4:13580230; added 50199 candidate reads
[M::seg] pass 1: read 400000 alignments, last at chr8:32597482; added 66488 candidate reads
[M::seg] pass 1: read 500000 alignments, last at chr11:21056174; added 82555 candidate reads
[M::seg] pass 1: read 600000 alignments, last at chr13:85023688; added 98829 candidate reads
[M::seg] pass 1: read 700000 alignments, last at chrX:120371153; added 115411 candidate reads
[M::seg] pass 1: read 800000 alignments, last at chr17:83341705; added 132316 candidate reads
[M::seg] pass 1: read 900000 alignments, last at chr7:122738108; added 147914 candidate reads
[M::seg] pass 1: read 1000000 alignments, last at chr9:117142127; added 163607 candidate reads
[M::seg] pass 1: read 1100000 alignments, last at chr8:89389020; added 180175 candidate reads
[M::seg] pass 1: read 1200000 alignments, last at chr2:136069232; added 195341 candidate reads
[M::seg] pass 1 done: read 1234782 alignments; added 200594 candidate reads (16.25% of alignments)
[M::seg] pass 2: read 100000 alignments, last at chr1:29284440
[M::seg] pass 2: read 200000 alignments, last at chr9:9941957
[M::seg] pass 2: read 300000 alignments, last at chr4:13580230
[M::seg] pass 2: read 400000 alignments, last at chr8:32597482
[M::seg] pass 2: read 500000 alignments, last at chr11:21056174
[M::seg] pass 2: read 600000 alignments, last at chr13:85023688
[M::seg] pass 2: read 700000 alignments, last at chrX:120371153
[M::seg] pass 2: read 800000 alignments, last at chr17:83341705
[M::seg] pass 2: read 900000 alignments, last at chr7:122738108
[M::seg] pass 2: read 1000000 alignments, last at chr9:117142127
[M::seg] pass 2: read 1100000 alignments, last at chr8:89389020
[M::seg] pass 2: read 1200000 alignments, last at chr2:136069232
[M::seg] pass 2: cleaning 200594 candidate reads
[M::seg] pass 2 done: read 1234782 alignments; kept 151510 candidate reads (12.27% of alignments)
Traceback (most recent call last):
File "./dip-c", line 122, in <module>
main()
File "./dip-c", line 40, in main
return_value = seg.seg(sys.argv[1:])
File "/Users/tarakshisode/dip-c/seg.py", line 129, in seg
for pileup_column in bam_file.pileup(snp_chr, snp_locus - 1, snp_locus):
File "pysam/libcalignmentfile.pyx", line 1327, in pysam.libcalignmentfile.AlignmentFile.pileup
ValueError: no index available for pileup
Am I doing something wrong??
Dear developer,
In Fig 2D of the Science paper, you showed the distances to the nuclear center in y-axis. I am just wondering how could I calculate the distance to the nuclear center. I did not find any clue after reading through the documentation. Thank you in advance.
Hi Tan,
Is there a way to obtain phased.snp.tsv file from mgp.v5.merged.snps_all.dbSNP142.vcf.gz file for the mentioned mouse strains? If so, how?
I am trying to expand my vcf.gz file but it fails to expand?!
Taraks-MacBook-Pro:downloads tarakshisode$ gunzip mgp.v5.merged.snps_all.dbSNP142.vcf.gz
gunzip: mgp.v5.merged.snps_all.dbSNP142.vcf.gz: unexpected end of file
gunzip: mgp.v5.merged.snps_all.dbSNP142.vcf.gz: uncompress failed
Hey Tan,
Is there a way to generate contact matrices based on chromosomes? Say if I want a matrix showing just chr1 or if I want to show chr1,chr2
Hi @tanlongzhi,
Hope you are doing good!
I have read the definition of single-cell chromatin Compartment in your paper, which is that
the single-cell chromatin compartment for each 1-Mb bin was calculated as the average CpG frequency of all other bins
contacting it, weighted by the number of contacts.
And I'm confused about it because I do not find relevant weighting steps in color2.py. How do you weigh it? Do you mean that e.g. if two bins have 3 contacts, the corresponding CpG frequency should be multiplied by 3, or other?
Any help again will be really great!
Dear @tanlongzhi,
How can I install Dip-C? I didn't find a readme installation instructions or a conda package.
Thank you.
Hi there,
Nice tools.
I am wondering is it possible to convert .3dg to pdb format?
Thanks.
Hi @tanlongzhi !
I'm looking to visualize the distance from a given allele and the periphery of the nucleus. However, I'm not sure how to find the radius of each nucleus. Taking the most extreme x, y and z value away from the center would likely overestimate the shape, as I imagine most nuclei are not perfectly spherical and may have some protrusions. Perhaps filtering out those extremes would be helpful. Perhaps calculating the radius of gyration of the nucleus would be a decent representation of the real radius. What are your thoughts? How would you go about finding the radius of each nucleus?
Thank you!!!
Hey @tanlongzhi ,
At present the pipeline requires the SNP VCF files to be given, but i was just curious whether using googles DeepVariant (a deep learning model for finding the variants) be useful or even help us find more phased legs??
I was thinking of implementing it. Let me know it makes sense to use it..
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.