smithlabcode / preseq Goto Github PK
View Code? Open in Web Editor NEWSoftware for predicting library complexity and genome coverage in high-throughput sequencing.
Home Page: https://preseq.readthedocs.io
License: GNU General Public License v3.0
Software for predicting library complexity and genome coverage in high-throughput sequencing.
Home Page: https://preseq.readthedocs.io
License: GNU General Public License v3.0

















































































Hi,
I am trying to build preseq on a linux machine. I've cloned the preseq repository and when I ran the 'make all' I was returned an error that there was no file matching 'smithlab_cpp//smithlab_os.o'. It seems like the the smithlab_cpp and preseqR directories have symbolic links to separate repos and did not contain any files when I cloned preseq. I've since populated these directories using the files from the respective repositories.
I'm not sure if this is the correct approach, but now when run make all, I am returned this error:

Thanks for any tips!
I have a bam file which I have filtered and sorted using samtools. When I run c_curve in paired-end mode, the command fails, claiming that the reads are unsorted. It is clear from the line numbers below that the reads are sorted correctly by samtools.
>samtools view -b -f 2 -q 4 original.bam > unsorted.bam
>samtools sort unsorted.bam final
>samtools index final.bam
>echo $?
0
>preseq c_curve -s 10000 -P -B final.bam
ERROR: reads unsorted in: final.bam
prev = chr1 1749306 1749357 HWI:1:X:1:2111:4871:48281 0 +
curr = chr1 1749275 1749326 HWI:1:X:1:2315:16167:50307 0 +
Increase seg_len if in paired end mode
>samtools view final.bam | grep -n HWI:1:X:1:2111:4871:48281
3126:HWI:1:X:1:2111:4871:48281 ... chr1 1749307 ...
3127:HWI:1:X:1:2111:4871:48281 ... chr1 1756855
>samtools view final.bam | grep -n HWI:1:X:1:2315:16167:50307
3125:HWI:1:X:1:2315:16167:50307 ... chr1 1749276 ...
3128:HWI:1:X:1:2315:16167:50307 ... chr1 1756860 ...
Hi,
I was wondering whether duplicates are considered or not for estimating the library complexity when they are marked with picard but still present in the bam file.
I read in the paper that you recommend to keep duplicates ("We therefore suggest that the genome coverage is predicted with duplicated included, but note that it can also be done with duplicates removed") but not sure if the program will filter them out based on the flag.
Thanks in advance,
Tamara
These should not exist, except under very rare circumstances. But there is no reason for them in comments.
The Makefile contains this line:
PROGS = preseq mincount_extrap mincount_c_curve saturation_extrap test_quadrature
However, there seem to be no targets to build all but preseq. To build preseq from sources I had to set PROGS=preseq. Otherwise the build would fail:
install: cannot stat ?mincount_extrap?: No such file or directory
install: cannot stat ?mincount_c_curve?: No such file or directory
install: cannot stat ?saturation_extrap?: No such file or directory
install: cannot stat ?test_quadrature?: No such file or directory
Makefile:105: recipe for target 'install' failed
make: *** [install] Error 1
None of the build rules create these four additional tools.
This part of the code is redundant because we set the internal RNG with srand, but then immediately define another RNG with a pre-defined value.
// setup rng
srand(time(0) + getpid());
std::mt19937 rng(seed);
Dear developers,
I am trying to install preseq in my mac.
I downloaded the preseq-master and I used make all.
I get this error:
make all
make: *** No rule to make target /Users/raffaelecalogero/Dropbox/calogero/delibero/single_cells/preseq-master/smithlab_cpp//smithlab_os.o', needed bypreseq'. Stop.
could please tell me where I can get smithlab_os.o
Cheers
Raf
@slzhao, I'm moving your issue to the preseq issues.
Does defect mode work when using 5M reads? Or just when using >50M?
The reasons for keeping preseqR separate no longer seem valid. Creating updates for CRAN seem no more difficult with preseqR as a submodule. What are the possible cons of having it simply a subdirectory here?
Needs to be cleaned up
Hi there.
I'm trying to run preseq 3.1.1 on a new Ubuntu 20.04.1 system through the command line. I have a prepared bam file, but when I run the program, I encounter this:
$ preseq lc_extrap PC1-PT.bam -B -P -e 2.1e9 -s 1e8 -seg_len 1000000000 -o PC1.preseq
ERROR: max count before zero is less than min required count (4) duplicates removed
Scouring the web for a solution, I look for a solution only to be met with an explanation here. But that doesn't appear accurate. It runs when I use a sam file as input of the same data.
I checked the help section and tried the -D option expecting some quality checks to be bypassed, but alas. Here I am. I want to know if there is any way to bypass this? I did not have this issue when working with version 2.0.3.
As an aside, and I can log this with another issue if it's a large task, one other item I noticed is that all the help pages seem to be formatted incorrectly, even the command line help. When I submit with the input file at the end of the options:
$ preseq lc_extrap -B -P -e 2.1e9 -s 1e8 -seg_len 1000000000 -o PC1.preseq PC1-PT.bam
ERROR: problem opening file: -B
I also did not encounter this issue in version 2.0.3.
Thanks.
I cloned preseq using git clone --recursive https://github.com/smithlabcode/preseq.git.
When I go into the preseq directory and use the make all command, I get the following error.
make: *** No rule to make target '/c/Users/cma83/Downloads/preseq/smithlab_cpp/smithlab_os.o', needed by 'preseq'. Stop.
Hi long time, no see. I've got an issue compiling from source on Mac OSv11.3.1. I get the following error:
preseq % make all
g++ -std=c++11 -Wall -O2 -c -o smithlab_cpp/smithlab_os.o smithlab_cpp/smithlab_os.cpp -Ismithlab_cpp
In file included from smithlab_cpp/smithlab_os.cpp:24:
/Library/Developer/CommandLineTools/SDKs/MacOSX.sdk/usr/include/c++/v1/cmath:321:9: error: no member named 'signbit' in the global namespace
using ::signbit;
~~^
/Library/Developer/CommandLineTools/SDKs/MacOSX.sdk/usr/include/c++/v1/cmath:322:9: error: no member named 'fpclassify' in the global namespace
using ::fpclassify;
~~^
/Library/Developer/CommandLineTools/SDKs/MacOSX.sdk/usr/include/c++/v1/cmath:323:9: error: no member named 'isfinite' in the global namespace; did you mean
'finite'?
using ::isfinite;
~~^
/usr/local/include/math.h:752:12: note: 'finite' declared here
extern int finite(double)
^
In file included from smithlab_cpp/smithlab_os.cpp:24:
/Library/Developer/CommandLineTools/SDKs/MacOSX.sdk/usr/include/c++/v1/cmath:324:9: error: no member named 'isinf' in the global namespace
using ::isinf;
~~^
/Library/Developer/CommandLineTools/SDKs/MacOSX.sdk/usr/include/c++/v1/cmath:325:9: error: no member named 'isnan' in the global namespace
using ::isnan;
~~^
/Library/Developer/CommandLineTools/SDKs/MacOSX.sdk/usr/include/c++/v1/cmath:326:9: error: no member named 'isnormal' in the global namespace
using ::isnormal;
~~^
/Library/Developer/CommandLineTools/SDKs/MacOSX.sdk/usr/include/c++/v1/cmath:327:7: error: no member named 'isgreater' in the global namespace; did you mean
'::std::greater'?
using ::isgreater;
^~
/Library/Developer/CommandLineTools/SDKs/MacOSX.sdk/usr/include/c++/v1/functional:738:29: note: '::std::greater' declared here
struct _LIBCPP_TEMPLATE_VIS greater : binary_function<_Tp, _Tp, bool>
^
In file included from smithlab_cpp/smithlab_os.cpp:24:
/Library/Developer/CommandLineTools/SDKs/MacOSX.sdk/usr/include/c++/v1/cmath:328:7: error: no member named 'isgreaterequal' in the global namespace; did you mean
'::std::greater_equal'?
using ::isgreaterequal;
^~
/Library/Developer/CommandLineTools/SDKs/MacOSX.sdk/usr/include/c++/v1/functional:767:29: note: '::std::greater_equal' declared here
struct _LIBCPP_TEMPLATE_VIS greater_equal : binary_function<_Tp, _Tp, bool>
^
In file included from smithlab_cpp/smithlab_os.cpp:24:
/Library/Developer/CommandLineTools/SDKs/MacOSX.sdk/usr/include/c++/v1/cmath:329:9: error: no member named 'isless' in the global namespace
using ::isless;
~~^
/Library/Developer/CommandLineTools/SDKs/MacOSX.sdk/usr/include/c++/v1/cmath:330:9: error: no member named 'islessequal' in the global namespace
using ::islessequal;
~~^
/Library/Developer/CommandLineTools/SDKs/MacOSX.sdk/usr/include/c++/v1/cmath:331:9: error: no member named 'islessgreater' in the global namespace
using ::islessgreater;
~~^
/Library/Developer/CommandLineTools/SDKs/MacOSX.sdk/usr/include/c++/v1/cmath:332:9: error: no member named 'isunordered' in the global namespace
using ::isunordered;
~~^
/Library/Developer/CommandLineTools/SDKs/MacOSX.sdk/usr/include/c++/v1/cmath:333:9: error: no member named 'isunordered' in the global namespace
using ::isunordered;
~~^
13 errors generated.
make[1]: *** [smithlab_cpp/smithlab_os.o] Error 1
make: *** [all] Error 2
I tried following the instructions from https://forums.swift.org/t/is-anyone-else-getting-this-error-when-building-the-compiler-from-master-on-macos/36113, specifically deleting /Library/Developer/CommandLineTools and running sudo xcode-select -s /Applications/Xcode.app. And.... same error.
I noticed that it's using g++ instead of clang. Is that part of the issue?
Thanks!
Hi, I'm trying to install preseq on a cluster where I don't have the permissions to install some applications to their default location. I unzipped the tar file and found that the ./preseq executable had the following persisting error:
preseq: error while loading shared libraries: libgsl.so.0: cannot open shared object file: No such file or directory
I set up gsl version 1.15 (I also tried version 2.6), configuring it to a certain prefix. Still same error. I also set up LD_LIBRARY_PATH to point to the same location as the prefix, exported the variable, and still got the same error.
Any ideas for why GSL isn't connecting to PreSeq? Thank you!
Hugo
Hi Tim, love your paper.
We ran into similar issues to #28 after converting a fastq to unaligned SAM/BAM with BBMap. I understand that coordinate-based pileup is the more efficient option for the UMI, but in a way it limits the applicability of preseq across NGS applications. In contrast, storing the sequences along with their counts in a hashmap would/could be prohibitively expensive.
So alternatively, you could take the approach of 2-bit encoding of the read like the python library kPAL. You could generate the vector from unique sequences instead of positions, which would be valuable for highly uniform libraries like 16S and other amplicon sequencing, where mapping is less relevant, accurate, or possible. Then your modeling/rarefaction process would probably be similar.
For some reasons preseq 3.0.2 and 3.0.1 do not compile on a Linux (debian 9) server.
Linux d4-app 4.15.0-45-generic #48-Ubuntu SMP Tue Jan 29 16:28:13 UTC 2019 x86_64 GNU/Linux
I've managed to resolve this by install version 3.0.0 but here are the logs for the failed compilation.
(env) tom@x86_64-conda_cos6-linux-gnu ➜ ~ tar -xvzf preseq-3.0.2.tar.gz
preseq-3.0.2/
preseq-3.0.2/install-sh
preseq-3.0.2/configure.ac
preseq-3.0.2/configure
preseq-3.0.2/preseqR/
preseq-3.0.2/config.h.in
preseq-3.0.2/depcomp
preseq-3.0.2/gpl-3.0.txt
preseq-3.0.2/missing
preseq-3.0.2/Makefile.am
preseq-3.0.2/README.md
preseq-3.0.2/m4/
preseq-3.0.2/data/
preseq-3.0.2/Makefile.in
preseq-3.0.2/aclocal.m4
preseq-3.0.2/src/
preseq-3.0.2/src/continued_fraction.cpp
preseq-3.0.2/src/load_data_for_complexity.hpp
preseq-3.0.2/src/moment_sequence.hpp
preseq-3.0.2/src/smithlab_cpp/
preseq-3.0.2/src/to-mr.cpp
preseq-3.0.2/src/continued_fraction.hpp
preseq-3.0.2/src/load_data_for_complexity.cpp
preseq-3.0.2/src/preseq.cpp
preseq-3.0.2/src/moment_sequence.cpp
preseq-3.0.2/src/smithlab_cpp/htslib_wrapper.hpp
preseq-3.0.2/src/smithlab_cpp/zlib_wrapper.hpp
preseq-3.0.2/src/smithlab_cpp/sim_utils.hpp
preseq-3.0.2/src/smithlab_cpp/install-sh
preseq-3.0.2/src/smithlab_cpp/configure.ac
preseq-3.0.2/src/smithlab_cpp/chromosome_utils.hpp
preseq-3.0.2/src/smithlab_cpp/configure
preseq-3.0.2/src/smithlab_cpp/ar-lib
preseq-3.0.2/src/smithlab_cpp/dna_four_bit.hpp
preseq-3.0.2/src/smithlab_cpp/MappedRead.hpp
preseq-3.0.2/src/smithlab_cpp/cigar_utils.hpp
preseq-3.0.2/src/smithlab_cpp/GenomicRegion.cpp
preseq-3.0.2/src/smithlab_cpp/smithlab_utils.hpp
preseq-3.0.2/src/smithlab_cpp/smithlab_os.cpp
preseq-3.0.2/src/smithlab_cpp/OptionParser.cpp
preseq-3.0.2/src/smithlab_cpp/QualityScore.cpp
preseq-3.0.2/src/smithlab_cpp/config.h.in
preseq-3.0.2/src/smithlab_cpp/bisulfite_utils.cpp
preseq-3.0.2/src/smithlab_cpp/depcomp
preseq-3.0.2/src/smithlab_cpp/missing
preseq-3.0.2/src/smithlab_cpp/Makefile.am
preseq-3.0.2/src/smithlab_cpp/README.md
preseq-3.0.2/src/smithlab_cpp/MappedRead.cpp
preseq-3.0.2/src/smithlab_cpp/GenomicRegion.hpp
preseq-3.0.2/src/smithlab_cpp/smithlab_os.hpp
preseq-3.0.2/src/smithlab_cpp/smithlab_utils.cpp
preseq-3.0.2/src/smithlab_cpp/OptionParser.hpp
preseq-3.0.2/src/smithlab_cpp/compile
preseq-3.0.2/src/smithlab_cpp/QualityScore.hpp
preseq-3.0.2/src/smithlab_cpp/bisulfite_utils.hpp
preseq-3.0.2/src/smithlab_cpp/m4/
preseq-3.0.2/src/smithlab_cpp/htslib_wrapper.cpp
preseq-3.0.2/src/smithlab_cpp/zlib_wrapper.cpp
preseq-3.0.2/src/smithlab_cpp/sim_utils.cpp
preseq-3.0.2/src/smithlab_cpp/Makefile.in
preseq-3.0.2/src/smithlab_cpp/chromosome_utils.cpp
preseq-3.0.2/src/smithlab_cpp/aclocal.m4
preseq-3.0.2/src/smithlab_cpp/dna_four_bit.cpp
preseq-3.0.2/src/smithlab_cpp/m4/ax_cxx_compile_stdcxx_11.m4
preseq-3.0.2/src/smithlab_cpp/m4/ax_cxx_compile_stdcxx.m4
preseq-3.0.2/data/SRR1003759_5M_subset.mr
preseq-3.0.2/data/SRR1106616_5M_subset.bam
preseq-3.0.2/data/additional_data.txt
preseq-3.0.2/data/SRR1301329_1M_read.txt
preseq-3.0.2/m4/ax_cxx_compile_stdcxx_11.m4
preseq-3.0.2/m4/ax_cxx_compile_stdcxx.m4
preseq-3.0.2/preseqR/man/
preseq-3.0.2/preseqR/R/
preseq-3.0.2/preseqR/NAMESPACE
preseq-3.0.2/preseqR/README.md
preseq-3.0.2/preseqR/DESCRIPTION
preseq-3.0.2/preseqR/.git
preseq-3.0.2/preseqR/inst/
preseq-3.0.2/preseqR/data/
preseq-3.0.2/preseqR/data/Dickens.txt
preseq-3.0.2/preseqR/data/SRR061157_k31.txt
preseq-3.0.2/preseqR/data/SRR611492_5M.txt
preseq-3.0.2/preseqR/data/WillButterfly.txt
preseq-3.0.2/preseqR/data/FisherButterfly.txt
preseq-3.0.2/preseqR/data/SRR1301329_base.txt
preseq-3.0.2/preseqR/data/SRR1301329_read.txt
preseq-3.0.2/preseqR/data/SRR611492.txt
preseq-3.0.2/preseqR/data/SRR1301329_1M_read.txt
preseq-3.0.2/preseqR/data/Twitter.txt
preseq-3.0.2/preseqR/data/Shakespeare.txt
preseq-3.0.2/preseqR/data/SRR1301329_1M_base.txt
preseq-3.0.2/preseqR/inst/CITATION
preseq-3.0.2/preseqR/R/kmer.R
preseq-3.0.2/preseqR/R/simulation.R
preseq-3.0.2/preseqR/R/sequencing.R
preseq-3.0.2/preseqR/R/rational_function.R
preseq-3.0.2/preseqR/R/compared_methods.R
preseq-3.0.2/preseqR/R/sample_coverage.R
preseq-3.0.2/preseqR/R/rSAC.R
preseq-3.0.2/preseqR/R/ztnb.R
preseq-3.0.2/preseqR/R/tools.R
preseq-3.0.2/preseqR/man/ds.rSAC.Rd
preseq-3.0.2/preseqR/man/ds.rSAC.bootsrap.Rd
preseq-3.0.2/preseqR/man/preseqR.ztnb.em.Rd
preseq-3.0.2/preseqR/man/SRR1301329_read.Rd
preseq-3.0.2/preseqR/man/preseqR.simu.hist.Rd
preseq-3.0.2/preseqR/man/Shakespeare.Rd
preseq-3.0.2/preseqR/man/SRR1301329_base.Rd
preseq-3.0.2/preseqR/man/FisherButterfly.Rd
preseq-3.0.2/preseqR/man/cs.rSAC.Rd
preseq-3.0.2/preseqR/man/Twitter.Rd
preseq-3.0.2/preseqR/man/preseqR.interpolate.rSAC.Rd
preseq-3.0.2/preseqR/man/preseqR.sample.cov.bootstrap.Rd
preseq-3.0.2/preseqR/man/fisher.alpha.Rd
preseq-3.0.2/preseqR/man/ztnb.rSAC.Rd
preseq-3.0.2/preseqR/man/preseqR.optimal.sequencing.Rd
preseq-3.0.2/preseqR/man/preseqR.package.Rd
preseq-3.0.2/preseqR/man/ztp.rSAC.Rd
preseq-3.0.2/preseqR/man/WillButterfly.Rd
preseq-3.0.2/preseqR/man/SRR611492_5M.Rd
preseq-3.0.2/preseqR/man/preseqR.rSAC.Rd
preseq-3.0.2/preseqR/man/kmer.frac.curve.Rd
preseq-3.0.2/preseqR/man/bbc.rSAC.Rd
preseq-3.0.2/preseqR/man/kmer.frac.curve.bootstrap.Rd
preseq-3.0.2/preseqR/man/SRR611492.Rd
preseq-3.0.2/preseqR/man/fisher.rSAC.Rd
preseq-3.0.2/preseqR/man/preseqR.sample.cov.Rd
preseq-3.0.2/preseqR/man/SRR1301329_1M_read.Rd
preseq-3.0.2/preseqR/man/preseqR.nonreplace.sampling.Rd
preseq-3.0.2/preseqR/man/preseqR.rSAC.bootstrap.Rd
preseq-3.0.2/preseqR/man/SRR1301329_1M_base.Rd
preseq-3.0.2/preseqR/man/preseqR.rSAC.sequencing.rmdup.Rd
preseq-3.0.2/preseqR/man/Dickens.Rd
preseq-3.0.2/preseqR/man/SRR061157_k31.Rd
(env) tom@x86_64-conda_cos6-linux-gnu ➜ ~ cd preseq-3.0.2
(env) tom@x86_64-conda_cos6-linux-gnu ➜ preseq-3.0.2 mkdir build
(env) tom@x86_64-conda_cos6-linux-gnu ➜ preseq-3.0.2 cd build
(env) tom@x86_64-conda_cos6-linux-gnu ➜ build ../configure --prefix=/home/tom/local/bin
checking for a BSD-compatible install... /usr/bin/install -c
checking whether build environment is sane... yes
checking for a thread-safe mkdir -p... /bin/mkdir -p
checking for gawk... gawk
checking whether make sets $(MAKE)... yes
checking whether make supports nested variables... yes
checking whether make supports nested variables... (cached) yes
checking for g++... g++
checking whether the C++ compiler works... yes
checking for C++ compiler default output file name... a.out
checking for suffix of executables...
checking whether we are cross compiling... no
checking for suffix of object files... o
checking whether we are using the GNU C++ compiler... yes
checking whether g++ accepts -g... yes
checking whether make supports the include directive... yes (GNU style)
checking dependency style of g++... gcc3
checking whether g++ supports C++11 features with -std=c++11... yes
checking for ranlib... ranlib
checking that generated files are newer than configure... done
configure: creating ./config.status
config.status: creating Makefile
config.status: creating config.h
config.status: executing depfiles commands
=== configuring in src/smithlab_cpp (/home/tom/preseq-3.0.2/build/src/smithlab_cpp)
configure: running /bin/sh ../../../src/smithlab_cpp/configure --disable-option-checking '--prefix=/home/tom/local/bin' 'LDFLAGS=-Wl,-O2 -Wl,--sort-common -Wl,--as-needed -Wl,-z,relro -Wl,-z,now -Wl,--disable-new-dtags -Wl,--gc-sections -Wl,-rpath,/home/tom/anaconda3/lib -Wl,-rpath-link,/home/tom/anaconda3/lib -L/home/tom/anaconda3/lib' --cache-file=/dev/null --srcdir=../../../src/smithlab_cpp
checking for a BSD-compatible install... /usr/bin/install -c
checking whether build environment is sane... yes
checking for a thread-safe mkdir -p... /bin/mkdir -p
checking for gawk... gawk
checking whether make sets $(MAKE)... yes
checking whether make supports nested variables... yes
checking whether make supports nested variables... (cached) yes
checking for g++... g++
checking whether the C++ compiler works... yes
checking for C++ compiler default output file name... a.out
checking for suffix of executables...
checking whether we are cross compiling... no
checking for suffix of object files... o
checking whether we are using the GNU C++ compiler... yes
checking whether g++ accepts -g... yes
checking whether make supports the include directive... yes (GNU style)
checking dependency style of g++... gcc3
checking whether g++ supports C++11 features with -std=c++11... yes
checking for gcc... gcc
checking whether we are using the GNU C compiler... yes
checking whether gcc accepts -g... yes
checking for gcc option to accept ISO C89... none needed
checking whether gcc understands -c and -o together... yes
checking dependency style of gcc... gcc3
checking for ar... ar
checking the archiver (ar) interface... ar
checking for ranlib... ranlib
checking for library containing z... no
checking that generated files are newer than configure... done
configure: creating ./config.status
config.status: creating Makefile
config.status: creating config.h
config.status: executing depfiles commands
(env) tom@x86_64-conda_cos6-linux-gnu ➜ build make
make all-recursive
make[1]: Entering directory '/home/tom/preseq-3.0.2/build'
Making all in src/smithlab_cpp
make[2]: Entering directory '/home/tom/preseq-3.0.2/build/src/smithlab_cpp'
make all-am
make[3]: Entering directory '/home/tom/preseq-3.0.2/build/src/smithlab_cpp'
CXX GenomicRegion.o
CXX MappedRead.o
CXX OptionParser.o
CXX QualityScore.o
CXX bisulfite_utils.o
CXX chromosome_utils.o
CXX sim_utils.o
CXX smithlab_os.o
CXX smithlab_utils.o
CXX zlib_wrapper.o
CXX dna_four_bit.o
AR libsmithlab_cpp.a
ar: `u' modifier ignored since `D' is the default (see `U')
make[3]: Leaving directory '/home/tom/preseq-3.0.2/build/src/smithlab_cpp'
make[2]: Leaving directory '/home/tom/preseq-3.0.2/build/src/smithlab_cpp'
make[2]: Entering directory '/home/tom/preseq-3.0.2/build'
CXX src/preseq.o
CXX src/continued_fraction.o
CXX src/load_data_for_complexity.o
CXX src/moment_sequence.o
In file included from ../src/moment_sequence.cpp:21:0:
../src/moment_sequence.hpp:28:1: error: ‘size_t’ does not name a type
size_t ensure_pos_def_mom_seq(std::vector<double> &moments,
^~~~~~
../src/moment_sequence.hpp:54:12: error: ‘size_t’ does not name a type
const size_t n_points,
^~~~~~
../src/moment_sequence.hpp:56:12: error: ‘size_t’ does not name a type
const size_t max_iter,
^~~~~~
../src/moment_sequence.cpp:373:1: error: prototype for ‘bool MomentSequence::Lower_quadrature_rules(bool, size_t, double, size_t, std::vector<double>&, std::vector<double>&)’ does not match any in class ‘MomentSequence’
MomentSequence::Lower_quadrature_rules(const bool VERBOSE,
^~~~~~~~~~~~~~
In file included from ../src/moment_sequence.cpp:21:0:
../src/moment_sequence.hpp:53:8: error: candidate is: bool MomentSequence::Lower_quadrature_rules(bool, int, double, int, std::vector<double>&, std::vector<double>&)
bool Lower_quadrature_rules(const bool VERBOSE,
^~~~~~~~~~~~~~~~~~~~~~
Makefile:515: recipe for target 'src/moment_sequence.o' failed
make[2]: *** [src/moment_sequence.o] Error 1
make[2]: Leaving directory '/home/tom/preseq-3.0.2/build'
Makefile:537: recipe for target 'all-recursive' failed
make[1]: *** [all-recursive] Error 1
make[1]: Leaving directory '/home/tom/preseq-3.0.2/build'
Makefile:377: recipe for target 'all' failed
make: *** [all] Error 2
(ega) tom@x86_64-conda_cos6-linux-gnu ➜ build make install
make[1]: Entering directory '/home/tom/preseq-3.0.2/build'
CXX src/moment_sequence.o
In file included from ../src/moment_sequence.cpp:21:0:
../src/moment_sequence.hpp:28:1: error: ‘size_t’ does not name a type
size_t ensure_pos_def_mom_seq(std::vector<double> &moments,
^~~~~~
../src/moment_sequence.hpp:54:12: error: ‘size_t’ does not name a type
const size_t n_points,
^~~~~~
../src/moment_sequence.hpp:56:12: error: ‘size_t’ does not name a type
const size_t max_iter,
^~~~~~
../src/moment_sequence.cpp:373:1: error: prototype for ‘bool MomentSequence::Lower_quadrature_rules(bool, size_t, double, size_t, std::vector<double>&, std::vector<double>&)’ does not match any in class ‘MomentSequence’
MomentSequence::Lower_quadrature_rules(const bool VERBOSE,
^~~~~~~~~~~~~~
In file included from ../src/moment_sequence.cpp:21:0:
../src/moment_sequence.hpp:53:8: error: candidate is: bool MomentSequence::Lower_quadrature_rules(bool, int, double, int, std::vector<double>&, std::vector<double>&)
bool Lower_quadrature_rules(const bool VERBOSE,
^~~~~~~~~~~~~~~~~~~~~~
Makefile:515: recipe for target 'src/moment_sequence.o' failed
make[1]: *** [src/moment_sequence.o] Error 1
make[1]: Leaving directory '/home/tom/preseq-3.0.2/build'
Makefile:537: recipe for target 'install-recursive' failed
make: *** [install-recursive] Error 1
I'm packaging preseq for Guix and would like to link against a binary version of samtools that is already available on the system rather than linking in samtools objects from a source checkout. Is this possible?
More specifically, samtools 1.1 and later comes with libbam.a. Would linking against this library be sufficient?
I wanted to note a discrepancy between sequencing data with and without duplicates. I know this is mentioned in the manuscript, Issue #31, and issue #24, but from what I can tell it seems that preseq is strongly affected by duplication, particularly as duplication rates increases. I unfortunately can't share the data presented below, but the experiment consisted on creating a single library from a single-cell amplification. This was sequenced on both a miniseq and a NovaSeq, both to around 5M reads. Notably, the NovaSeq has a known higher duplication rate. I aligned with BWA-MEM to the human genome hg38, and converted both the raw alignment and the deduplicated alignment with bam2mr. In our hands, the miniseq has ~0.4% duplication rate, and the novaseq has ~8% duplication rate.
This is the result of preseq's gc_extrap on the miniseq/novaseq raw/deduplicated data.

Again, I am sorry that I can't share the data (at least publicly, we might be able to with an NDA with my company). Have you run into anything like this before? It seems contrary to the figure presented in the original paper, so perhaps this is some weird characteristic of novaseq data. I have seen this now across about 20 of these miniseq/novaseq pairs.
Thanks, any input would be appreciated!
Receive this error when attempting to run Preseq:
Command error:
BAM_INPUT
ERROR: malformed line in SAM format:
NB501293:85:HGGFMBGXY:4:12504:17802:7321 1:N:0:TGACCA 129 1 dna_sm:chromosome chromosome:GRCh37:1:1:249250621:1 REF 130060 17 1L1
Pair ended RNA-seq data, stranded, aligned with BBMap, sorted using Sambamba.
Any ideas?
oss declared on line 303, error is as follows:
load_data_for_complexity.cpp:317:7: error: use of
undeclared identifier 'oss'
oss << "locations unsorted in: " << ...
^
load_data_for_complexity.cpp:320:31: error: use of
undeclared identifier 'oss'
throw SMITHLABException(oss.str());
Probably a good time to do this.
It looks like you've added a version 2.0.3. Can you tag that in github so the bioconda recipe can be updated? We prefer to use officially tagged releases there.
I noticed it giving negative output sometimes: tail -1 test.txt 999900000000.0 1063084.0 -528316.0 4777585.5
I put it in a for loop, and it seems to happen pretty often.
Hi, I am trying to troubleshoot an issue with using the to-mr executable to covert a bam file generated with bwa mem. I am using preseq in a docker container build from the following dockerfile:
FROM ubuntu:20.04
ARG VER="3.1.1"
RUN apt-get update && apt-get install wget autoconf automake make gcc perl zlib1g-dev libbz2-dev liblzma-dev libcurl4-gnutls-dev libssl-dev g++ -y
RUN wget https://github.com/samtools/htslib/releases/download/1.9/htslib-1.9.tar .bz2 && \
tar -vxjf htslib-1.9.tar.bz2 && \
cd htslib-1.9 && \
make && \
make install
ENV LD_LIBRARY_PATH /htslib-1.9/
RUN wget https://github.com/smithlabcode/preseq/releases/download/v${VER}/pres\
eq-${VER}.tar.gz &&\
tar xzf preseq-${VER}.tar.gz &&\
cd preseq-${VER} &&\
mkdir build && cd build &&\
../configure --enable-hts &&\
make &&\
make install
I ran this on a file containing about 5M reads, and got the error below. for reproducing the error, I took a subset of the BAM, and it is attached.
I ran gc_extrap as follows and got the following error:
$ to-mr -o /data/sample.mr /data/sample.bam -L 10000 -v
[input file: /data/sample.bam]
[output file: /data/sample.mr]
$ preseq gc_extrap -o /data/sample.curve /data/sample.mr -v
LOADING READS
MAPPED READ FORMAT
ERROR: bad line in MappedRead file: (NULL) 0 0 X 0 +
Looking through the MR file, it seems that the following reads are causing the problem:
A00758:111:HL2FCDSXY:4:2369:32416:19554 131 chr1 16788 0 75M = 17055 268 ACTAGGGAAGAAGGTGTG
TGACCAGGGAGGTCCCCGGCCCAGCTCCCATCCCAGAACCCAGCTCACCTACCTTGA FFFFFFFFFF:FFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFF
FFFFFFFFFFFFFFFFF NM:i:1 MD:Z:0T74 MC:Z:75M AS:i:74 XS:i:74 XA:Z:chr1,+187310,75M,1;chr2,-113596574,74
M1S,1;chr16,+16472,1S74M,1;chr15,-101974098,75M,2;chr9,+16900,1S74M,1;
A00758:111:HL2FCDSXY:4:2369:32416:19554 67 chr1 17055 0 75M = 16788 -268 CCTGCAAGATTAGGCAGG
GACATGTGAGAGGTGACAGGGACCTGCAGGGGCAGCCAACAAGACCTTGTGTGCACC FF:FFFFF,FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
FFFFFFFFFFFFFFFFF NM:i:0 MD:Z:75 MC:Z:75M AS:i:75 XS:i:75 XA:Z:chrX,-156022938,75M,0;chr16,+16738,75M,0;chr2
,-113596307,75M,0;chr12,+17170,75M,0;chr9,+17166,75M,0;chr15,-101973831,75M,0;chr1,+187577,75M,0;chr12_GL877875v1_alt,+717
0,75M,0;
Running with bam2mr warns of several segment length issues, but results in an output file containing the passing reads:
~/bin/preseq_v2.0/bam2mr -o sample.old.mr sample.bam -v -seg_len 100000 2> old.log
Those files are in the attached zip as well.
Is there something wrong with my BAM file? I have been using preseq 2.0.3 for a while (and bam2mr), and am trying to update to 3.1.1. Converting the BAM to BED with bedtools's bedToBam and running preseq results in no errors.
Thanks in advance!
Apologies if I'm being slow here, but is there a version history for preseq anywhere? The homepage talks about a compiled version 0.1.0 but a source code download version 1.0.1..
It would be great if releases could be dated and tagged so that we can be sure that we're running the latest (stable) code. Thanks!
It looks like there were some recent header file name changes that may be causing this. The compiler throws the error:
./src/load_data_for_complexity.cpp:184:10: fatal error: htslib_wrapper.hpp: No such file or directory
#include <htslib_wrapper.hpp>
Indeed, it seems htslib_wrapper.hpp (and .cpp) is missing, although the file htslib_wrapper_deprecated.hpp is present. What's strange is that the preseq repo contains a submodule pointing to a commit that does have both files present, so I'm not sure why it's missing or what else might be going on here.
Any suggestions would be much appreciated!
Hi,
Thanks a lot for this great tool! When preseq is predict the unique number of reads based on the complexity of reads, if there is UMI on one or both end of the reads, will it be aware of the existence of UMI?
thanks,
Yaping
hello
i tried git clone
git clone --recursive git://github.com/smithlabcode/preseq.git
then
make all SAMTOOLS_DIR=/Users/christos/Desktop/samtools
and got
Volumes/SD/preseq/smithlab_cpp/SAM.hpp:28:10: fatal error:
'sam.h' file not found
#include "sam.h"
^
1 error generated.
make: *** [load_data_for_complexity.o] Error 1
I then tried without the samtools options
Make all
make: gcc-4.8”: No such file or directory
make: *** [/Volumes/SD/preseq/samtools//sam.o] Error 1
then after downloading the mac image
eduroam-int-dhcp-97-58-98:preseq_osx_v2.0 christos$ ./preseq c_curve -o output.txt input.sort.bed
dyld: Library not loaded: /usr/local/lib/libgsl.0.dylib
Referenced from: /Users/christos/Downloads/preseq_osx_v2.0/./preseq
Reason: image not found
Trace/BPT trap: 5
any suggestions welcome!
For the lc_extrap function, the documentation says you can use BED or BAM as inputs. No mention of WALT mr files. But mr files seem to work just fine. So can I confirm this: Are mr files a valid input for preseq lc_extrap?
I have tried to 'make all' the preseq directory using the gcc 8.2 compiler and gsl 2.5 but get errors.
preseq.cpp:(.text+0x2d25): undefined reference to strip_path(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >)' /tmp/ccqhKVFD.o: In function gc_extrap(int, char const**)':
preseq.cpp:(.text+0x540c): undefined reference to strip_path(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >)' /tmp/ccqhKVFD.o: In function c_curve(int, char const**)':
preseq.cpp:(.text+0x7b48): undefined reference to strip_path(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >)' /tmp/ccqhKVFD.o: In function bound_pop(int, char const**)':
preseq.cpp:(.text+0x94ef): undefined reference to strip_path(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >)' /ihome/rseal/cma83/preseq/smithlab_cpp/OptionParser.o: In function Option::format_option_description[abi:cxx11](unsigned long, bool) const':
OptionParser.cpp:(.text+0x103b): undefined reference to smithlab::split_whitespace(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> > const&, std::vector<std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, std::allocator<std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> > > >&)' /ihome/rseal/cma83/preseq/smithlab_cpp/OptionParser.o: In function Option::parse_config_file(std::vector<std::__cxx11::basic_string<char, std::char_traits, std::allocator >, std::allocator<std::__cxx11::basic_string<char, std::char_traits, std::allocator > > >&)':
OptionParser.cpp:(.text+0x17fa): undefined reference to smithlab::split(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, char const*, bool)' OptionParser.cpp:(.text+0x182d): undefined reference to smithlab::strip(std::__cxx11::basic_string<char, std::char_traits, std::allocator > const&)'
OptionParser.cpp:(.text+0x1881): undefined reference to smithlab::strip(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> > const&)' /ihome/rseal/cma83/preseq/smithlab_cpp/OptionParser.o: In function OptionParser::about_messageabi:cxx11 const':
OptionParser.cpp:(.text+0x3f94): undefined reference to smithlab::split_whitespace(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> > const&, std::vector<std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, std::allocator<std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> > > >&)' /ihome/rseal/cma83/preseq/smithlab_cpp/MappedRead.o: In function GenomicRegion::GenomicRegion()':
MappedRead.cpp:(.text._ZN13GenomicRegionC2Ev[_ZN13GenomicRegionC5Ev]+0x38): undefined reference to GenomicRegion::assign_chrom(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> > const&)' /ihome/rseal/cma83/preseq/smithlab_cpp/MappedRead.o: In function GenomicRegion::GenomicRegion(std::__cxx11::basic_string<char, std::char_traits, std::allocator >, unsigned long, unsigned long, std::__cxx11::basic_string<char, std::char_traits, std::allocator >, float, char)':
MappedRead.cpp:(.text._ZN13GenomicRegionC2ENSt7__cxx1112basic_stringIcSt11char_traitsIcESaIcEEEmmS5_fc[_ZN13GenomicRegionC5ENSt7__cxx1112basic_stringIcSt11char_traitsIcESaIcEEEmmS5_fc]+0x2f): undefined reference to GenomicRegion::assign_chrom(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> > const&)' /ihome/rseal/cma83/preseq/smithlab_cpp/MappedRead.o: In function std::__cxx11::basic_ostringstream<char, std::char_traits, std::allocator >& operator<< <std::__cxx11::basic_ostringstream<char, std::char_traits, std::allocator > >(std::__cxx11::basic_ostringstream<char, std::char_traits, std::allocator >&, GenomicRegion const&)':
MappedRead.cpp:(.text._ZlsINSt7__cxx1119basic_ostringstreamIcSt11char_traitsIcESaIcEEEERT_S7_RK13GenomicRegion[_ZlsINSt7__cxx1119basic_ostringstreamIcSt11char_traitsIcESaIcEEEERT_S7_RK13GenomicRegion]+0x20): undefined reference to GenomicRegion::tostring[abi:cxx11]() const' load_data_for_complexity.o: In function GenomicRegion::get_chromabi:cxx11 const':
load_data_for_complexity.cpp:(.text._ZNK13GenomicRegion9get_chromB5cxx11Ev[_ZNK13GenomicRegion9get_chromB5cxx11Ev]+0x20): undefined reference to GenomicRegion::retrieve_chrom[abi:cxx11](unsigned int)' load_data_for_complexity.o: In function std::ostream& operator<< std::ostream(std::ostream&, GenomicRegion const&)':
load_data_for_complexity.cpp:(.text._ZlsISoERT_S1_RK13GenomicRegion[_ZlsISoERT_S1_RK13GenomicRegion]+0x20): undefined reference to `GenomicRegion::tostringabi:cxx11 const'
collect2: error: ld returned 1 exit status
make: *** [preseq] Error 1
Is this my fault??

The web page (and github README) seem to imply that the current version in github is >=2.0.0. However, the version string in preseq.cpp is 1.1.2
Running preseq c_curve on a paired end sorted bam gives me the following error:
Singularity> preseq c_curve -o out.nomerge.curve -P -B ex640_P_mapped.sorted.bam
ERROR: reads unsorted in: ex640_P_mapped.sorted.bam
prev = scaffold_1 112694 112734 A00709:254:HTK7WDSX2:3:2625:13612:18865 0 +
curr = scaffold_1 111263 111307 A00709:254:HTK7WDSX2:3:2576:30544:8046 0 +
Increase seg_len if in paired end mode
c_curve runs fine if I don't use the -P option.
strangely, when I look into the bam, I don't see reads at the positions mentioned in the error: scaffold_1:112694 or scaffold_1:111263.
I also resorted the bam using samtools sort but still got the same error.
Occurs on install from github and v1.0.2 source tarball.
Error points to line 37 in preseq.cpp.
I presume this will present a similar problem for lines 38 and 39, as well.
Not that it should matter:
OS: Ubuntu 14.04 (Bio-Linux 8)
gcc: 4.8.2
gsl: 1.16
fatal: reference is not a tree: 0f0e9e6d71fe72104ec7242c9cf8c701df6bc69b
Unable to checkout '0f0e9e6d71fe72104ec7242c9cf8c701df6bc69b' in submodule path 'smithlab_cpp'
Any ideas?
[...]
aits<char>, std::allocator<char> >, bool, unsigned long&)'
preseq.cpp:(.text+0xa258): undefined reference to `OptionParser::add_opt(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, char, std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, bool, double&)'
preseq.cpp:(.text+0xa35a): undefined reference to `OptionParser::add_opt(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, char, std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, bool, unsigned long&)'
preseq.cpp:(.text+0xa468): undefined reference to `OptionParser::add_opt(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, char, std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, bool, double&)'
preseq.cpp:(.text+0xa540): undefined reference to `OptionParser::add_opt(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, char, std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, bool, bool&)'
preseq.cpp:(.text+0xa609): undefined reference to `OptionParser::add_opt(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, char, std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, bool, bool&)'
preseq.cpp:(.text+0xa6d2): undefined reference to `OptionParser::add_opt(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, char, std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, bool, bool&)'
preseq.cpp:(.text+0xa79b): undefined reference to `OptionParser::add_opt(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, char, std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, bool, bool&)'
preseq.cpp:(.text+0xa864): undefined reference to `OptionParser::add_opt(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, char, std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, bool, bool&)'
preseq.cpp:(.text+0xa957): undefined reference to `OptionParser::add_opt(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, char, std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, bool, unsigned long&)'
preseq.cpp:(.text+0xaa2f): undefined reference to `OptionParser::add_opt(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, char, std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, bool, bool&)'
preseq.cpp:(.text+0xaaaa): undefined reference to `OptionParser::parse(int, char const**, std::vector<std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, std::allocator<std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> > > >&)'
preseq.cpp:(.text+0xaaef): undefined reference to `_ZNK12OptionParser12help_messageB5cxx11Ev'
preseq.cpp:(.text+0xab68): undefined reference to `_ZNK12OptionParser13about_messageB5cxx11Ev'
preseq.cpp:(.text+0xabe1): undefined reference to `_ZNK12OptionParser22option_missing_messageB5cxx11Ev'
preseq.cpp:(.text+0xac5a): undefined reference to `_ZNK12OptionParser12help_messageB5cxx11Ev'
load_data_for_complexity.o: In function `load_counts_BAM_se(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> > const&, std::vector<double, std::allocator<double> >&)':
load_data_for_complexity.cpp:(.text+0x7d7): undefined reference to `SAMReader::SAMReader(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >)'
load_data_for_complexity.o: In function `load_counts_BAM_pe(bool, std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> > const&, unsigned long, unsigned long, unsigned long&, unsigned long&, std::vector<double, std::allocator<double> >&)':
load_data_for_complexity.cpp:(.text+0x14b1): undefined reference to `SAMReader::SAMReader(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >)'
load_data_for_complexity.o: In function `GenomicRegion::GenomicRegion(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, unsigned long, unsigned long, std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> >, float, char)':
load_data_for_complexity.cpp:(.text._ZN13GenomicRegionC2ENSt7__cxx1112basic_stringIcSt11char_traitsIcESaIcEEEmmS5_fc[_ZN13GenomicRegionC5ENSt7__cxx1112basic_stringIcSt11char_traitsIcESaIcEEEmmS5_fc]+0x2f): undefined reference to `GenomicRegion::assign_chrom(std::__cxx11::basic_string<char, std::char_traits<char>, std::allocator<char> > const&)'
load_data_for_complexity.o: In function `_ZNK13GenomicRegion9get_chromB5cxx11Ev':
load_data_for_complexity.cpp:(.text._ZNK13GenomicRegion9get_chromB5cxx11Ev[_ZNK13GenomicRegion9get_chromB5cxx11Ev]+0x20): undefined reference to `_ZN13GenomicRegion14retrieve_chromB5cxx11Ej'
collect2: error: ld returned 1 exit status
make: *** [preseq] Error 1
[rocks-av@rocks1 preseq]$ g++ --version
g++ (GCC) 5.2.0
Copyright (C) 2015 Free Software Foundation, Inc.
This is free software; see the source for copying conditions. There is NO
warranty; not even for MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE.
Looks like lots of old debug code, including some formerly hard-coded filenames, are still apparent in some of the source files.
The latest release tarball is not self-contained. While it is good that preseq does not bundle smithlab_cpp, it is not easy to build preseq as there has not been an official release of the smithlab_cpp library.
Just unpacking the tarball and building preseq results in an error:
make: *** No rule to make target '/tmp/nix-build-preseq-2.0.drv-0/preseq-2.0/smithlab_cpp//smithlab_os.o', needed by 'preseq'. Stop.
In my latest version of the preseq package for GNU Guix I'm first building a prelease of smithlab_cpp and then set SMITHLAB_CPP to the lib output of that package; I also modify the INCLUDEDIRS variable such that the installed headers of the smithlab_cpp package can be found.
If there will be a release for smithlab_cpp soon, it would be nicer if preseq could be linked against that library directly.
I just downloaded the v3.1.1 release and tried to install both with and without htslib. Running make failed, giving the following errors:
In file included from ../src/moment_sequence.cpp:21:0:
../src/moment_sequence.hpp:28:1: error: ‘size_t’ does not name a type
size_t ensure_pos_def_mom_seq(std::vector<double> &moments,
^~~~~~
../src/moment_sequence.hpp:54:12: error: ‘size_t’ does not name a type
const size_t n_points,
^~~~~~
../src/moment_sequence.hpp:56:12: error: ‘size_t’ does not name a type
const size_t max_iter,
^~~~~~
../src/moment_sequence.cpp:373:1: error: prototype for ‘bool MomentSequence::Lower_quadrature_rules(bool, size_t, double, size_t, std::vector<double>&, std::vector<double>&)’ does not match any in class ‘MomentSequence’
MomentSequence::Lower_quadrature_rules(const bool VERBOSE,
^~~~~~~~~~~~~~
In file included from ../src/moment_sequence.cpp:21:0:
../src/moment_sequence.hpp:53:8: error: candidate is: bool MomentSequence::Lower_quadrature_rules(bool, int, double, int, std::vector<double>&, std::vector<double>&)
bool Lower_quadrature_rules(const bool VERBOSE,
^~~~~~~~~~~~~~~~~~~~~~
I attempted to create a pull request with my fix but I couldn't figure out how to integrate the preseqR and smithlab_cpp submodules into my fork.
I was able to fix the errors and build/install successfully by just adding the following include line to src/moment_sequence.hpp:
#include <cstddef>
Could you please tag one or more of your commits as a release? Maybe corresponding to points in time that you've released preseqR to CRAN.
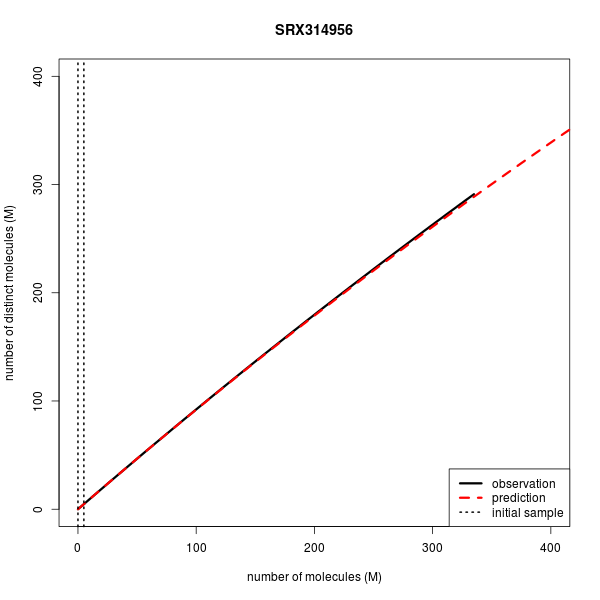
I have had a look around the Preseq website as I am interested in implementing it as part of our ChIP-Seq workflow, and also to stimulate some discussions with lab members regarding the re-sequencing of existing ChIP libraries. I came across the complexity challenge pages, and was surprised to see the plot for the WGBS library:
http://smithlabresearch.org/wp-content/uploads/SRX314956-1.png
Is this a typical behaviour for WGBS data? It seems that it would be nearly impossible to ever reach saturation in this type of experiment...
Anyway, thanks for this tool, I think it will be very helpful in order to assess library quality prior to performing more sequencing runs.
Problem
I am getting different values for complexity output and future yield when generating the outputs using BAM or BED file. I might be doing something wrong so please look at the workflow below:
BAM
samtools sort fly_aligned.bam -o bam/fly_aligned.sorted.bam
preseq c_curve -o complexity_output.txt -B fly_aligned.sorted.bam
preseq lc_extrap -o future_yield.txt -B fly_aligned.sorted.bam
BED
bedtools bamtobed -bed12 -i fly_aligned.bam > bed/fly_aligned.bed
sort -k 1,1 -k 2,2n -k 3,3n -k 6,6 fly_aligned.bed > fly_aligned.sorted.bed
preseq c_curve -o complexity_output.txt fly_aligned.sorted.bed
preseq lc_extrap -o future_yield.txt fly_aligned.sorted.bed
R Plots
fy_bed = read.table("future_yield.txt", header=TRUE) # bed future yield
sub_fy_bed <- fy_bed[1:2]
fy_bam = read.table("future_yield.txt", header=TRUE) # bam future yield
sub_fy_bam <- fy_bam[1:2]
plot(sub_fy_bed, type="p", col="red", pch=3, lwd=1, main="Future Yield", xlab="Total Reads", ylab="Expected Distinct")
points(sub_fy_bam, type="p", col="green", pch=1, lwd=1)
legend(5e+09, 7.0e+06, c("BED", "BAM"), lty=c(3, 1), lwd=c(1,1),col=c("red", "green"))

co_bed = read.table("complexity_output.txt", header=TRUE)
co_bam = read.table("complexity_output.txt", header=TRUE)
sub_co_bed <- co_bed[1:2]
sub_co_bam <- co_bam[1:2]
plot(co_bed, type="p", col="red", pch=3, lwd=1, main="Complexity Plot", xlab="Total Reads", ylab="Distinct Reads")
points(co_bam, type="p", col="green", pch=1, lwd=1)
legend(5e+09, 7.0e+06, c("BED", "BAM"), lty=c(3, 1), lwd=c(1,1),col=c("red", "green"))

They look very different and I was wondering why the BAM workflow is producing different results from the BED workflow.
Any ideas?
Behram
Hi,
I was plotting complexity curves for some single-cells experiments and as you can see in the plot below, for some cases, the total number of bases covered decreases when increasing sequencing depth. How is this possible?
This is the command line I run:
bam2mr -v -o ${SAMPLE}.mr ${SAMPLE}.sorted.bam preseq gc_extrap -v -o ${SAMPLE}_inferredcov.txt ${SAMPLE}.mr

Hi there,
I am trying to install v3.1.2 to our Ubuntu 16.04 system, get an error below:
g++ -std=c++11 -DHAVE_CONFIG_H -I. -I/opt/apps/htslib/1.3.1/htslib -O3 -MT htslib_wrapper_deprecated.o -MD -MP -MF .deps/htslib_wrapper_deprecated.Tpo -c -o htslib_wrapper_deprecated.o htslib_wrapper_deprecated.cpp
In file included from htslib_wrapper_deprecated.cpp:25:0:
htslib_wrapper_deprecated.hpp:31:24: fatal error: htslib/sam.h: No such file or directory
while the file sam.h is there:
$ ls /opt/apps/htslib/1.3.1/htslib
bgzf.h cram.h faidx.h hfile.h hts_defs.h hts.h kbitset.h kfunc.h khash.h khash_str2int.h klist.h knetfile.h kseq.h ksort.h kstring.h regidx.h sam.h synced_bcf_reader.h tbx.h vcf.h vcf_sweep.h vcfutils.h
can anyone suggest how to fix this? thanks in advance.
Hi,
My histogram file contains 0 0 in the first line, e.g.:
0 0
1 60833379
2 26578973
3 9192783
4 2756185
5 767258
...
In the example you have provided in the manual I saw that your example starts with 1. I tried both and got the exact same results for lc_extrap. I have been using the histogram with 0's for a while now. Do you think it is necessary to repeat all these preseq runs with deleting the first line of histogram, or can I assume I would get the same output?
Thank you,
We have a SAM.hpp and SAM.cpp file, presumably for some wrapper, but when I try to build the code it complains about no sam.h. Is this intended behavior?
Examples including
mapper = "genera"
should not exist. This is not C, it's C++, and we don't need to code like that.
I think the following appearing in headers works because versions of gcc supported them by extensions, not because it's standard C++. Current versions of gcc are warning against this:
// included in header in class definition
static const double SEARCH_MAX_VAL = 100;
static const double SEARCH_STEP_SIZE = 0.05;
The above should be declared in the class definition, but then defined outside, preferably in compiled code (the *.cpp).
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.
{kind=link}