版本Version:v1.21
更新时间Update:2024/5/17
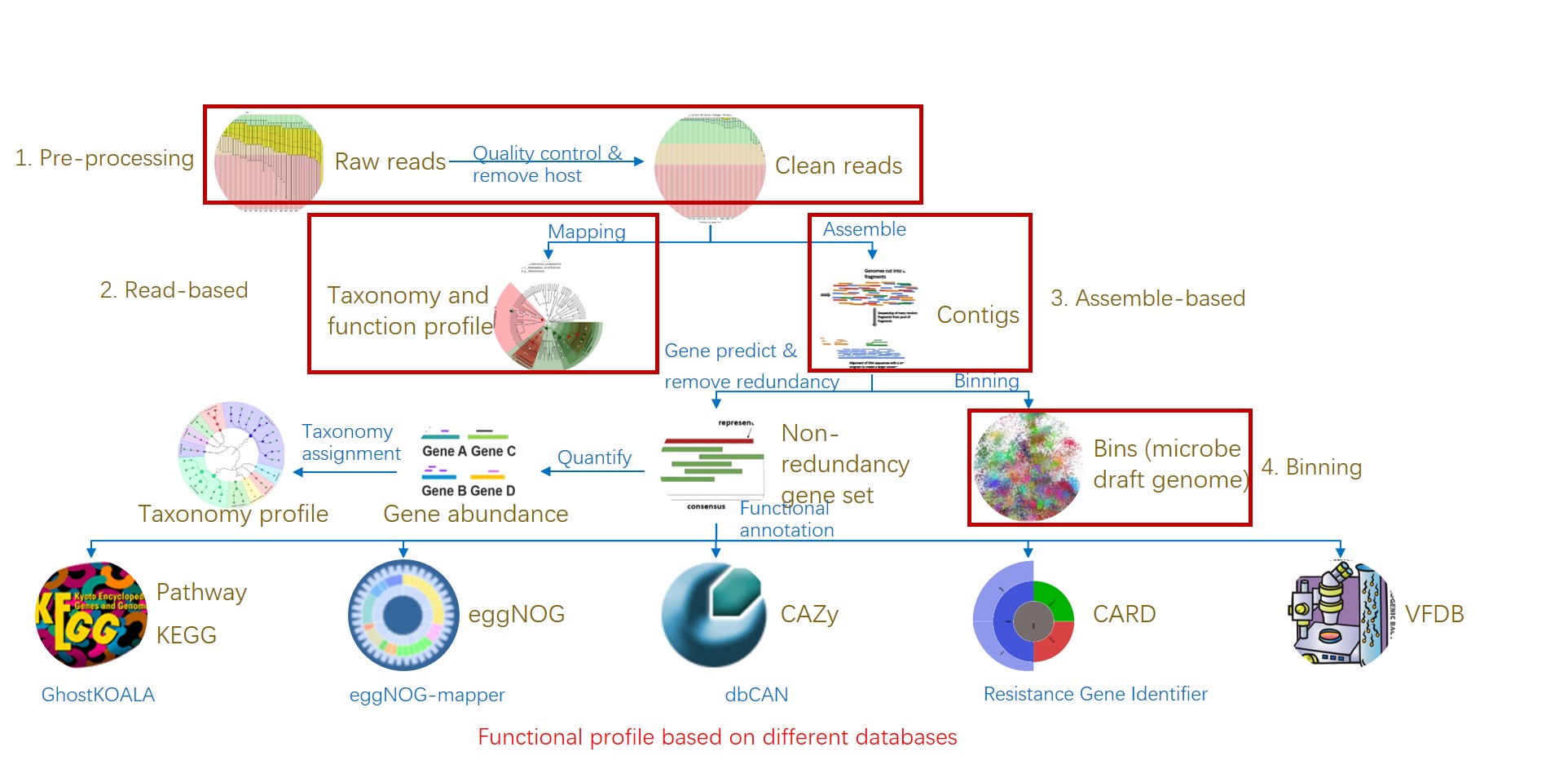
图1. 易宏基因组的工作流程:分析宏基因组测序从原始数据到物种和功能组成表.
Figure 1. EasyMetagenome Pipeline from raw data to taxonomic & functional table for analyzing metagenomic sequencing.
- 0Install.sh:软件和数据库安装Software and database installation
- 1Pipeline.sh:分析流程Analysis pipeline
- 2StatPlot.sh:统计和可视化Statistics and visualization
Shell脚本(.sh)兼容Markdown格式,可使用有道云笔记、VSCode等工具中Markdown格式查看,有目录导航更方便浏览和阅读。
The Shell scripts are compatible with the Markdown format, which can be viewed in the Markdown editor, such as YoudaoCloudNotes, VSCode, and the menu navigation is more convenient for browsing and reading.
各文档附录部分为常见问题,供参考。
The appendices of each document are frequently asked questions for reference.
在64位版本Linux系统,如Ubuntu 20.04+ / CentOS 7.7+,按代码文件0,1,2顺序逐个运行
In 64-bit version Linux system, such as Ubuntu 20.04+ / CentOS 7.7+, run step by step according to scripts 0, 1, 2.
在终端命令行,或RStudio的Terminal环境下使用,可以有道云笔记中显示代码目录,方便预览大纲
Used in the terminal command line or RStudio's terminal environment. You can display the code menu in Youdaoyun Notes, which is convenient for previewing the outline
If used this script, please cited
使用此脚本,请引用下文:
Yong-Xin Liu, Lei Chen, Tengfei Ma, Xiaofang Li, Maosheng Zheng, Xin Zhou, Liang Chen, Xubo Qian, Jiao Xi, Hongye Lu, Huiluo Cao, Xiaoya Ma, Bian Bian, Pengfan Zhang, Jiqiu Wu, Ren-You Gan, Baolei Jia, Linyang Sun, Zhicheng Ju, Yunyun Gao, Tao Wen, Tong Chen. 2023. EasyAmplicon: An easy-to-use, open-source, reproducible, and community-based pipeline for amplicon data analysis in microbiome research. iMeta 2: e83. https://doi.org/10.1002/imt2.83
Yong-Xin Liu, Yuan Qin, Tong Chen, Meiping Lu, Xubo Qian, Xiaoxuan Guo, Yang Bai. 2021. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein & Cell 12: 315-330. https://doi.org/10.1007/s13238-020-00724-8 (Highly Cited)
Copyright 2016-2023 Yong-Xin Liu [email protected], Tong Chen [email protected]













































































































