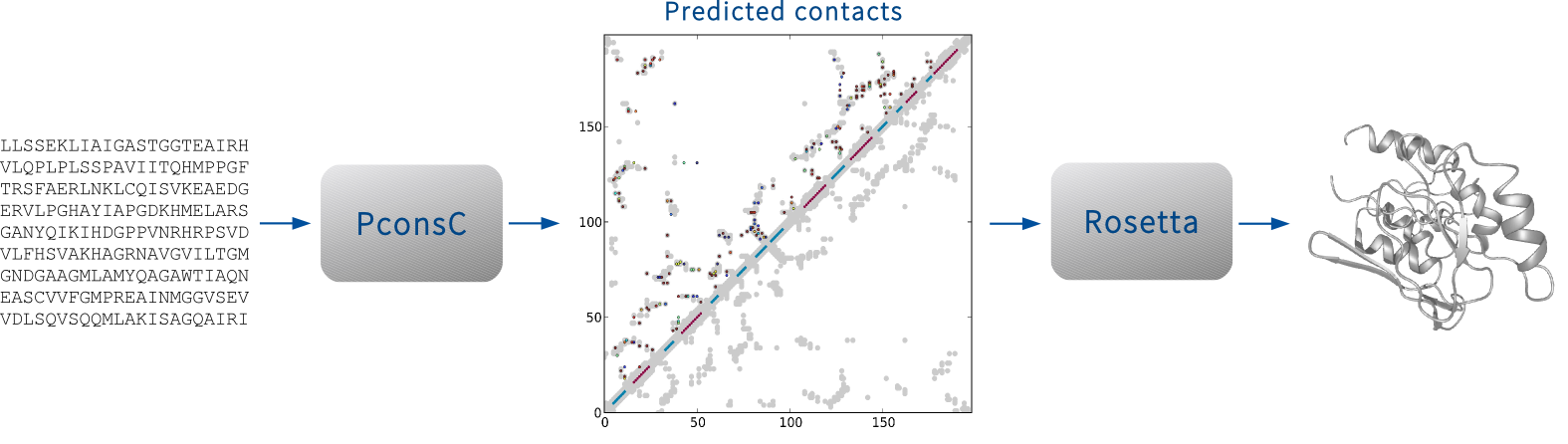
A pipeline for protein folding using predicted contacts from PconsC and a Rosetta folding protocol.
You find supplementary data, such as protein IDs, sequences, native and predicted structures, predicted contacts at the bottom of the release page.
- Input: fasta file containing one protein sequence
- Prepare input for PconsC
- Contact prediction with PconsC
- Prepare input for Rosetta folding
- Rosetta folding
- Extract and relax structures with lowest Rosetta energy
- Output: the predicted contact map (also as a plot) and the top-ranked structural model(s) relaxed and non-relaxed
- Rosetta v3.5 or weekly built
- Jackhmmer from HMMER v3.0 or higher
- HHblits from HHsuite v2.0.16
- PSICOV v1.11
- plmDCA asymmetric
- either MATLAB v8.1 or higher
- or MATLAB Compiler Runtime (MCR) v8.1
MATLAB is needed to run plmDCA. However, if MATLAB is not available you can also use a compiled version of plmDCA. For the compiled version to run you need to provide a path to MCR.
Make sure all dependencies are working correctly and adjust the paths in localconfig.py.
To run the full pipeline use:
./pcons_fold.py [-c n_cores] [-n n_decoys] [-m n_models]
[-f factor] [--norelax] [--nohoms]
hhblits_database jackhmmer_database sequence_file
- Required:
hhblits_databaseandjackhmmer_databaseare paths to the databases used by HHblits and Jackhmmersequence_fileis the path to the input protein sequence in FASTA format (only single sequences).
- Optional:
n_coresspecifies the number of cores to use during computation (default: number of available cores).n_decoysspecifies the number of decoy structures generated by Rosetta (default: 2000, see publication).n_modelsis the number of top-ranked models being extracted and eventually relaxed in the end (default: 10).factordetermines the number of constraints used to fold the protein, which is:factor* length_of_the_input_sequence (default: 1.0).norelaxis a flag that supresses relaxation of the final models. This can be used to quickly extract structures in the end.nohomsis a flag that ensures that homologous structures are excluded from fragment picking. This is only useful in test cases if the model quality needs to be evaluated with a known structure.
You can also run PconsC contact prediction independently with this command:
./pconsc/predict_all.py [-c cores] hhblits_database jackhmmer_database sequence_file
And then fold the protein according to given predicted contacts with the following commands:
./folding/rosetta/prepare_input.py [-f factor] [--nohoms] sequence_file contact_map
./folding/rosetta/fold.py [-c n_cores] [-n n_decoys] sequence_file rosetta_constraintfile
./folding/rosetta/extract.py [-c n_cores] [-m n_models] [--norelax] number_of_extracted_structures
The first script generates the file (pconsc_output)-(factor).constraints which is then used by Rosetta in the next step with rosetta_constraintfile.