VIA is a single-cell Trajectory Inference method that offers topology construction, pseudotimes, automated terminal state prediction and automated plotting of temporal gene dynamics along lineages. VIA combines lazy-teleporting random walks and Monte-Carlo Markov Chain simulations to overcome common challenges such as 1) accurate terminal state and lineage inference, 2) ability to capture combination of cyclic, disconnected and tree-like structures, 3) scalability in feature and sample space. 4) Generalizability to multi-omic analysis. In addition to transcriptomic data, VIA works on scATAC-seq, flow and imaging cytometry data.
Please refer to our readthedocs and our paper for more details. .

Installations and tutorials available on readthedocs with step-by-step code for real and simulated datasets.
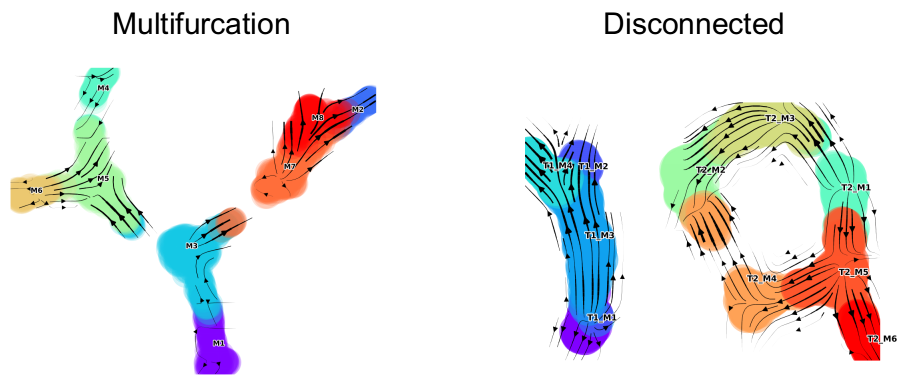
✳️ You can start with the The tutorial/Notebook for multifurcating data which shows a step-by-step use case. ✳️
scATAC-seq dataset of Human Hematopoiesis represented by VIA graphs (click image to open interactive graph)

Please visit our readthedocs for the latest tutorials.
| notebook | details | dataset | reference |
|---|---|---|---|
| Multifurcation: Starter Tutorial | 4-leaf simulation | 4-leaf | DynToy |
| Disconnected | disconnected simulation | Disconnected | DynToy |
| Human Embryoid | 16,825 ESCs | EB scRNA-seq and embedding | Moon et al. (2019) |
| FACED image-based | 2036 MCF7 cells in cell cycle | MCF7 FACED | in-house data |
| scATAC-seq Hematopoiesis | Human hematopoiesis | scATAC-seq | Buenrostro et al. (2018) |
| scRNA-seq Hematopoiesis | Human hematopoiesis (5780 cells) | CD34 scRNA-seq | Setty et al. (2019) |
The examples below show how to run VIA on generic connected and disconnected data using wrapper functions and serve as a check for your installation. For more detailed guidelines on running VIA and plotting the results, please use the Notebooks. We also highlight a few difference in calling VIA when using Windows versus Linux. The data for the Jupyter Notebooks and Examples are available in the Datasets folder (smaller files) with larger datasets here.
A test script is available for some of the different datasets, please change the foldername accordingly to the folder containing relevant data files
- 1.a Toy Data (multifurcation) Multifurcation Jupyter NB
- 1.b Toy Data (disconnected) Disconnected Jupyter NB
- 2.a Human Embryoid Bodies (wrapper function for testing VIA)
- 2.b Human Embryoid Bodies (Configuring VIA) EB Jupyter NB
- 3.a General input data formatting and wrapper function
- 3.b General disconnected trajectories wrapper function
Two examples toy datasets with annotations generated using DynToy are provided. For the step-by-step code within these wrappers, please see the corresponding Jupyter NBs.
Multifurcating toy dataset with interactive graph


Refer to the Jupiter Notebooks to plot these fine-grained vector fields of the sc-trajectories even when there is no RNA-velocity available.
All examples are shown according to Linux OS, small modifications are required to run on a Windows OS (see below)
import pyVIA.core as via
# ensure the data and label files are in csv format when you download/save them
#multifurcation
#the root is automatically set to root_user = 'M1'
via.main_Toy(ncomps=10, knn=30,dataset='Toy3', random_seed=2,foldername = ".../Trajectory/Datasets/") #multifurcation
#disconnected trajectory
# The root is automatically set as a list root_user = ['T1_M1', 'T2_M1'] # e.g. T2_M3 is a cell belonging to the 3rd Milestone (M3) of the second Trajectory (T2)
via.main_Toy(ncomps=10, knn=30,dataset='Toy4',random_seed=2,foldername =".../Trajectory/Datasets/") #2 disconnected trajectories
Windows requires minor modifications in calling the code due to the way multiprocessing works in Windows compared to Linux:
#when running from an IDE you need to call the function in the following way to ensure the parallel processing works:
import os
import pyVIA.core as via
f= os.path.join(r'C:\Users\...\Documents'+'\\')
def main():
via.main_Toy(ncomps=10, knn=30,dataset='Toy3', random_seed=2,foldername= f)
if __name__ =='__main__':
main()
#when running directly from terminal:
import os
import pyVIA.core as via
f= os.path.join(r'C:\Users\...\Documents'+'\\')
via.main_Toy(ncomps=10, knn=30,dataset='Toy3', random_seed=2,foldername= f)
Save the Raw data matrix as 'EBdata.mat'. The cells in this file have been filtered for too small/large libraries by Moon et al. 2019.
Save the phate embedding which is required to run Example 2.a (Or use another 2D embedding of your choice)
The function main_EB_clean() is a wrapper function which preprocesses the cells (normalized by library size, sqrt transformation). It then calls VIA to: plot the pseudotimes, terminal states, lineage pathways and gene-clustermap. The visualization method used in this function is PHATE.
#runtime on single core, 8GB RAM 64bit Windows is about 8-10 minutes. This can be lowered by reducing the number of MCMC simulations (num_mcmc_simulations).
import pyVIA.core as via
#Windows example path for folder where EBdata.mat is saved: f= os.path.join(r'C:\Users\...\Documents'+'\\')
via.main_EB_clean(ncomps=30, knn=20, v0_random_seed=24, foldername = f) # Most reasonable parameters of ncomps (10-200) and knn (15-50) work well
If you require more control of the parameters/outputs or choice of embedding, then use the following code which is otherwise wrapped within main_EB_clean(). (See the Jupyter Notebook for detailed code and outputs). Expected runtime will be around 1-2 minutes using 5 cores, or ~8-10 on "normal" laptop. .

Datasets and labels used in this example are provided in Datasets.
# Read the two files:
# 1) The first file contains 200PCs of the Bcell filtered and normalized data for the first 5000 HVG.
# 2) The second file contains raw count data for marker genes
data = pd.read_csv('./Bcell_200PCs.csv')
data_genes = pd.read_csv('./Bcell_markergenes.csv')
data_genes = data_genes.drop(['cell'], axis=1)
true_label = data['time_hour']
data = data.drop(['cell', 'time_hour'], axis=1)
adata = sc.AnnData(data_genes)
adata.obsm['X_pca'] = data.values
# use UMAP or PHate to obtain embedding that is used for single-cell level visualization
embedding = umap.UMAP(random_state=42, n_neighbors=15, init='random').fit_transform(data.values[:, 0:5])
# list marker genes or genes of interest if known in advance. otherwise marker_genes = []
marker_genes = ['Igll1', 'Myc', 'Slc7a5', 'Ldha', 'Foxo1', 'Lig4', 'Sp7'] # irf4 down-up
# call VIA. We identify an early (suitable) start cell root = [42]. Can also set an arbitrary value
via.via_wrapper(adata, true_label, embedding, knn=10, ncomps=20, jac_std_global=0.15, root=[42], dataset='',
random_seed=1,v0_toobig=0.3, v1_toobig=0.1, marker_genes=marker_genes)
import scanpy as sc
import pandas as pd
#foldername corresponds to the location where you have saved the Toy Disconnected data (shown in example 2)
#Read in the data and labels
df_counts = pd.read_csv(foldername + "toy_disconnected_M9_n1000d1000.csv", 'rt', delimiter=",")
df_ids = pd.read_csv(foldername + "toy_disconnected_M9_n1000d1000_ids.csv", 'rt', delimiter=",")
# Make AnnData object for wrapper function to read-in data and do PCA
df_ids['cell_id_num'] = [int(s[1::]) for s in df_ids['cell_id']]
df_counts = df_counts.drop('Unnamed: 0', 1)
df_ids = df_ids.sort_values(by=['cell_id_num'])
df_ids = df_ids.reset_index(drop=True)
true_label = df_ids['group_id']
adata_counts = sc.AnnData(df_counts, obs=df_ids)
sc.tl.pca(adata_counts, svd_solver='arpack', n_comps=10)
#Since there are 2 disconnected trajectories, we provide 2 arbitrary roots (start cells).If there are more disconnected paths, then VIA arbitrarily selects roots. #The root can also just be arbitrarily set as [1] and VIA can detect how many additional roots it must add
# The root can also be provided as a cell type level label corresponding to the groups present in "true_label", in this case the dataset must be set to 'group'
via.via_wrapper_disconnected(adata_counts, true_label, embedding=adata_counts.obsm['X_pca'][:, 0:2], root=[1,1], preserve_disconnected=True, knn=30, ncomps=10,cluster_graph_pruning_std = 1)
#in the case of connected data (i.e. only 1 graph component. e.g. Toy Data Multifurcating) then the wrapper function from example 3.a can be used:
via.via_wrapper(adata_counts, true_label, embedding= adata_counts.obsm['X_pca'][:,0:2], root=[1], knn=30, ncomps=10,cluster_graph_pruning_std = 1)
We recommend setting up a new conda environment. You can use the examples below, the Jupyter notebooks and/or the test script to make sure your installation works as expected.
conda create --name ViaEnv python=3.7
pip install pyVIA // tested on linux Ubuntu 16.04 and Windows 10
This usually tries to install hnswlib, produces an error and automatically corrects itself by first installing pybind11 followed by hnswlib. To get a smoother installation, consider installing in the following order after creating a new conda environment:
pip install pybind11
pip install hnswlib
pip install pyVIA
git clone https://github.com/ShobiStassen/VIA.git
python3 setup.py install // cd into the directory of the cloned VIA folder containing setup.py and issue this command
The pie-chart cluster-graph plot does not render correctly for MACs for the time-being. All other outputs are as expected.
conda create --name ViaEnv python=3.7
pip install pybind11
conda install -c conda-forge hnswlib
pip install pyVIA
If the pip install doesn't work, it usually suffices to first install all the requirements (using pip) and subsequently install VIA (also using pip). Note that on Windows if you do not have Visual C++ (required for hnswlib) you can install using this link .
pip install pybind11, hnswlib, igraph, leidenalg>=0.7.0, umap-learn, numpy>=1.17, scipy, pandas>=0.25, sklearn, termcolor, pygam, phate, matplotlib,scanpy
pip install pyVIA
| Input Parameter for class VIA | Description |
|---|---|
data |
(numpy.ndarray) n_samples x n_features. When using via_wrapper(), data is ANNdata object that has a PCA object adata.obsm['X_pca'][:, 0:ncomps] and ncomps is the number of components that will be used. |
true_label |
(list) 'ground truth' annotations or placeholder |
knn |
(optional, default = 30) number of K-Nearest Neighbors for HNSWlib KNN graph |
root_user |
root_user should be provided as a list containing roots corresponding to index (row number in cell matrix) of root cell. For most trajectories this is of the form [53] where 53 is the index of a sensible root cell, for multiple disconnected trajectories an arbitrary list of cells can be provided [1,506,1100], otherwise VIA arbitratily chooses cells. If the root cells of disconnected trajectories are known in advance, then the cells should be annotated with similar syntax to that of Example Dataset in Disconnected Toy Example 1b. |
dist_std_local |
(optional, default = 1) local pruning threshold for PARC clustering stage: the number of standard deviations above the mean minkowski distance between neighbors of a given node. the higher the parameter, the more edges are retained |
jac_std_global |
(optional, default = 0.15) global level graph pruning for PARC clustering stage. This threshold can also be set as the number of standard deviations below the network's mean-jaccard-weighted edges. 0.1-1 provide reasonable pruning. higher value means less pruning. e.g. a value of 0.15 means all edges that are above mean(edgeweight)-0.15*std(edge-weights) are retained. We find both 0.15 and 'median' to yield good results resulting in pruning away ~ 50-60% edges |
too_big_factor |
(optional, default = 0.4) if a cluster exceeds this share of the entire cell population, then the PARC will be run on the large cluster |
x_lazy |
(optional, default = 0.95) 1-x = probability of staying in same node (lazy). Values between 0.9-0.99 are reasonable |
alpha_teleport |
(optional, default = 0.99) 1-alpha is probability of jumping. Values between 0.95-0.99 are reasonable unless prior knowledge of teleportation |
distance |
(optional, default = 'l2' euclidean) 'ip','cosine' |
random_seed |
(optional, default = 42) The random seed to pass to Leiden |
pseudotime_threshold_TS |
(optional, default = 30) Percentile threshold for potential node to qualify as Terminal State |
resolution_parameter |
(optional, default = 1) Uses ModuliartyVP and RBConfigurationVertexPartition |
preserve_disconnected_after_pruning |
(optional, default = False) Cluster-graph pruning can occasionally cause fragmentation that can be repaired (by setting to True) by retaining select edges. |
cluster_graph_pruning_std |
(optional, default =0.15) Usually set to the same value as the PARC clustering level of jac_std_global. To retain more connectivity in the graph underlying the trajectory computations, increase the value |
visual_cluster_graph_pruning |
(optional, default = 0.15) Usually set to the same value as the PARC clustering level of jac_std_global. This does not impact computation of terminal states, pseudotime or lineage likelihoods. It controls the number of edges plotted for visual effect |
num_sim_branch_probability |
(optional), default = 500. Number of MCMCs run per terminal state. This can be safely reduced to 100 when computational resources are limited |
As shown in the examples, VIA is built to run on a single or double iteration. For extremely large datasets (>1M cells), a single pass is favourable. For mid-size it can be useful to run the double-pass mode which is shown in Example 2b and 3a,b. In the first pass VIA performs a coarser clustering which is useful for capturing terminal states. To increase the resolution of the pseudotime, lineage likelihood and gene trends, we re-run VIA in a second pass which produces a finer cluster-graph and transfers the lineages (terminal states) obtained in the first pass into the second pass. We therefore provide the second pass with the terminal_states obtained in the first pass. To speed up the computation, we also pass the original HNSW KNN graph. The following parameters are used for second passes.
| Input Parameter | Description |
|---|---|
is_coarse |
(optional, default = True) If running VIA in two steps, for the second fine-grained, set to "False' |
via_coarse |
(optional, default = None) If a second fine-grained iteration of VIA is run using the terminal states, roots and single-cell graph obtained in the first coarse-pass of VIA, then via_coarse = v0 (the VIA object from first iteration) |
| Attributes | Description |
|---|---|
labels |
(list) length n_samples of corresponding cluster labels |
edgelist_maxout |
(list) used to draw trajectories on the 2D embedding |
single_cell_pt_markov |
(list) computed pseudotime |
single_cell_bp |
(array) computed single cell branch probabilities (lineage likelihoods). n_cells x n_terminal states. The columns each correspond to a terminal state, in the same order presented in the'terminal_clusters' attribute |
terminal clusters |
(list) terminal clusters found by VIA |
super_cluster_labels |
Set this to v0.labels (clustering output of first pass "v0") |
super_terminal_cells |
super_terminal_cells = via.get_loc_terminal_states(v0, data) |
full_neighbor_array |
full_neighbor_array=v0.full_neighbor_array. KNN graph from first pass of via - neighbor array |
full_distance_array |
full_distance_array=v0.full_distance_array. KNN graph from first pass of via - edge weights |
ig_full_graph |
ig_full_graph=v0.ig_full_graph igraph of the KNN graph from first pass of via |
csr_array_locally_pruned |
csr_array_locally_pruned=v0.csr_array_locally_pruned. CSR matrix of the locally pruned KNN graph |

