alona (yes, spelled with a lowercase 'a') is a scientific analysis software for high-dimensional gene expression data from single cells, so-called scRNA-seq data. The typical input dataset to alona contains thousands of columns and rows. Each column represents one biological cell and rows are measured features (in our case, genes). alona automates almost all steps in the analysis of the input data.
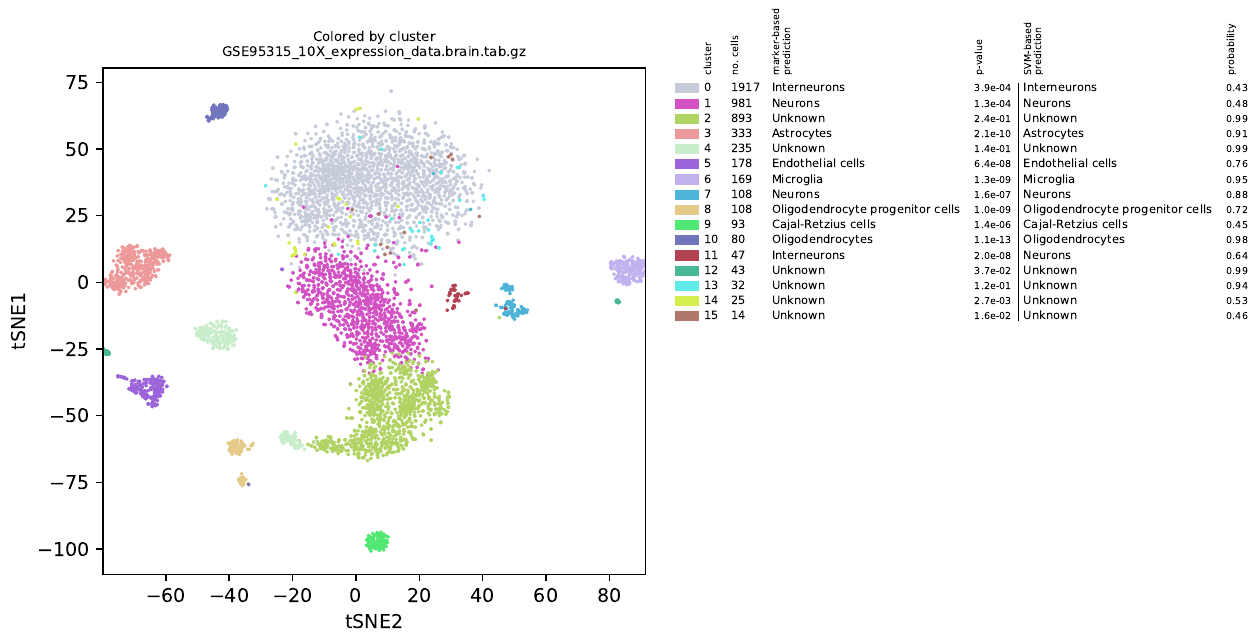
alona is a Python-based software for analysis of scientific data generated using a technology called single cell RNA sequencing (scRNA-seq) (1). alona is command-line centered and performs normalization, quality control, clustering, cell type annotation and visualization. alona integrates many state of the art algorithms in scRNA-seq analysis to a single convenient tool. In comparison with other scRNA-seq analysis pipelines, alona is designed as a command-line tool and not primarily as an importable library. Nevertheless, alona is modular and specific functions can be imported into existing Python projects. The goal ofalona is to facilitate fast and consistent single cell data analysis without requiring typing new code; at the same time, alona is written in portable and easily modifiable Python code, which makes it easy to adopt to new projects. Running alona to analyze scRNA-seq data is simple, fast and requires very little tweaking, as most parameters have sensible defaults. The clustering method used in alona is similar to the R package called Seurat; i.e., first computing k nearest neighbors in PCA space, followed by generating a shared nearest neighbor graph. alona uses the Leiden algorithm (an improved version of the Louvain algorithm) to identify tightly connected communities from the graph.
alona also exists as a parallel cloud-based service (2), which contains a subset of the features of the standalone version.
It depends on your goals. The goal of alona is to enable fast exploratory analysis of new datasets by automating as many steps as possible. alona generates data tables which can easily be loaded into your analysis toolkit of choice, where more in-depth statistical analysis can take place.
- Linux or MacOS (should in principle work on Windows too, but it has not been tested)
- Python (version >= 3.6)
alona relies heavily on numpy, pandas, matplotlib, scipy and others. Complete list of dependencies (missing dependencies are installed if pip3 is used for installation, see below): click, matplotlib, numpy, pandas, scipy, scikit-learn, leidenalg, umap-learn, statsmodels, igraph, and seaborn.
The fastest way to install alona is to first clone the GitHub repository and then use pip to install it. pip is a package manager for Python. If you don't have pip installed, it can be installed by the following command on Debian-based systems (e.g. Ubuntu):
sudo apt-get install python3-pipThen simply...
# Clone the repository
git clone https://github.com/oscar-franzen/alona/
# Enter the directory
cd alona
# Install the package
python3 -m pip install . --user
# Clean
cd ..
rm -rfv alonaalona works on scRNA-seq data from any organism. However, cell type prediction methods only work with human and mouse data. If your organism is not mouse and human, use the flag --species other to indicate this.
Pre-processing (mapping, read counting, etc) of FASTQ files is not a step included in alona, because processing of FASTQ files is usually performed on high performance clusters whereas the analysis can be performed on a laptop or desktop computer. We provide a short tutorial on how to preprocess FASTQ files, please see here.
The input file is a single gene expression matrix in plain text format. The header of the matrix are barcodes and the first column are gene symbols. Fields should be separated by tabs, commas or spaces (but not a mix). The file can be compressed with zip, gzip or bzip2. In addition, data can also be in Matrix Market format (a format popular in NCBI GEO), consisting of three files (one file for the actual data values, a second file for barcodes and a third file for gene symbols), which must be bundled together in a tar file (can be compressed with gzip or not).
ERCC spikes can be included and will be automatically detected and handled. Make sure ERCC "genes" are labeled with the prefix ERCC_ or ERCC-.
Here is one example of calling the pipeline using the data set GSM3689776.
python3 -m alona \
--species human \
--embedding tSNE \
--hvg_n 1000 \
--leiden_res 0.1 \
--output ~/Temp/test \
--header yes \
--minexpgenes 0.001 \
--remove_mito yes GSM3689776_mouse_10X_matrix.txt.gzHere is an example of the directory structure and files created by alona in the output directory test.
rand[~/alona/python-alona/test]> tree
.
├── csvs
│ ├── clusters_leiden.csv
│ ├── CTA_RANK_F
│ │ ├── cell_type_pred_best.txt
│ │ └── cell_type_pred_full_table.txt
│ ├── embeddings.csv
│ ├── highly_variable_genes.csv
│ ├── Mahalanobis.csv
│ ├── median_exp.csv
│ ├── pca.csv
│ ├── snn_graph.csv
│ └── SVM
│ ├── SVM_cell_type_pred_best.txt
│ └── SVM_cell_type_pred_full_table.txt
├── input.mat
├── input.mat.C
├── normdata_ERCC.joblib
├── normdata.joblib
├── plots
│ ├── 2d_plot_tSNE.pdf
│ ├── barplot_ge.pdf
│ └── barplot_rrc.pdf
├── settings.txt
└── unmappable.txt
4 directories, 20 files
rand[~/]> python3 -m alona --help
Usage: alona.py [OPTIONS] FILENAME
Options:
-o, --output TEXT Specify name of output directory
-df, --dataformat [raw|rpkm|log2]
How the input data have been processed. For
example, if data are raw read counts, then
select "raw". [default: raw]
-mr, --minreads INTEGER Minimum number of reads per cell to keep the
cell. [default: 1000]
-mg, --minexpgenes FLOAT Pre-filter the data matrix and remove genes
according to this threshold. Can be
specified either as a fraction of all cells
or as an an integer (translates to the
absolute number of cells that at minimum
must express the gene). [default: 0.01]
--qc_auto BOOLEAN Automatic filtering of low quality cells.
[default: True]
--mrnafull Data come from a full-length protocol, such
as SMART-seq2. [default: False]
--exclude_gene TEXT Remove any gene matching this regular
expression.
-d, --delimiter [auto|tab|space]
Data delimiter. The character used to
separate data values. The default setting is
to autodetect this character. [default:
auto]
-h, --header [auto|yes|no] Data has a header line. The default setting
is to autodetect if a header is present or
not. [default: auto]
-m, --remove_mito [yes|no] Remove mitochondrial genes from analysis
[default: no]
--hvg [seurat|Brennecke2013|scran|Chen2016|M3Drop_smartseq2|M3Drop_UMI]
Method to use for identifying highly
variable genes. [default: seurat]
--hvg_n INTEGER Number of top highly variable genes to use.
[default: 1000]
--pca [irlb|regular] PCA method to use. [default: irlb]
--pca_n INTEGER Number of PCA components to use. [default:
75]
--nn_k INTEGER k in the nearest neighbour search.
[default: 10]
--prune_snn FLOAT Threshold for pruning the SNN graph, i.e.
the edges with lower value (Jaccard index)
than this will be removed. Set to 0 to
disable pruning. Increasing this value will
result in fewer edges in the graph.
[default: 0.067]
--leiden_partition [RBERVertexPartition|ModularityVertexPartition]
Partitioning algorithm to use. Can be
RBERVertexPartition or
ModularityVertexPartition. [default:
RBERVertexPartition]
--leiden_res FLOAT Resolution parameter for the Leiden
algorithm (0-1). [default: 0.8]
--ignore_small_clusters INTEGER
Ignore clusters with fewer or equal to N
cells. [default: 10]
--annotations PATH An optional file containing gene
descriptions. The first column in this file
must be gene identifiers and the second
column is any string. The two columns should
be separated by a tab character. No header
is allowed. Gene symbols must match gene
symbols of the input data matrix.
--custom_clustering PATH An optional file containing a pre-generated
clustering. This option can be used if
clustering has already been performed
externally. The file should contain two
columns, delimited by a tab character,
without header. The first column should
contain cell identifiers and the second
column should contain the cluster.
--embedding [tSNE|UMAP] Method used for data projection. Can be
either tSNE or UMAP. [default: tSNE]
--perplexity INTEGER The perplexity parameter in the t-SNE
algorithm. [default: 30]
-s, --species [human|mouse|other]
Species your data comes from. [default:
mouse]
--dark_bg Use dark background in scatter plots.
[default: False]
--de_direction [any|up|down] Direction for differential gene expression
analysis.
--add_celltypes TEXT Add markers for these additional cell types
to the heatmap plot. Separate multiple cell
types with commas.
--overlay_genes TEXT Generate scatter plots in 2d space (using
method specified by --embedding), where gene
expression is overlaid on cells. Specify
multiple genes by comma separating gene
symbols.
--highlight_specific_cells TEXT
Specific cells can be highlighted in scatter
plots in 2d space (using method specified by
--embedding). Specify multiple cells by
comma separating cell identifiers (usually
barcodes).
--violin_top INTEGER Generate violin plots for the specified
number of top expressed genes per cluster.
[default: 10]
--timestamp Add timestamp label to plots. [default:
False]
-lf, --logfile TEXT Name of log file. Set to /dev/null if you
want to disable logging to a file.
[default: alona.log]
-ll, --loglevel [regular|debug]
Set how much runtime information is written
to the log file. [default: regular]
-n, --nologo Hide the logo.
--seed INTEGER Set seed to get reproducible results.
--version Display version number.
--help Show this message and exit.
| option | detailed description |
|---|---|
-out, --output [TEXT] |
Specify name of output directory. If this is not given then a directory with the format: alona_out_N will be created, where N is a 8 letter random string, in the current working directory. |
-df, --dataformat [raw|rpkm|log2] |
Specifies how the input data has been normalized. There are currently three options: raw means input data are raw read counts (alona will take care of normalization steps); rpkm means input data are normalized as RPKM but not logarithmized and alona will not perform any more normalization except for loging; log2 means that input data have been normalized and logarithmized and alona will not perform these steps. Default: raw |
--minexpgenes [float|int], -mg |
Pre-filter the data matrix and remove genes according to this threshold. Can be specified either as a fraction of all cells or as an an integer (translates to the absolute number of cells that at minimum must express the gene). |
--mrnafull |
Data come from a full-length protocol, such as SMART-seq2. This option is important if data represent full mRNAs. Drop-seq/10X and similar protocols sequence the ENDS of an mRNA, it is therefore not necessary to normalize for gene LENGTH. However, if we sequence the complete mRNA then we must also normalize measurements for the length of the gene, since longer genes have more mapped reads. If this option is not set, then cell type prediction may give unexpected results when analyzing full-length mRNA data. Default: False |
--hvg [method] |
Method to use for identifying highly variable genes, must be one of: seurat, Brennecke2013, scran, Chen2016, M3Drop_smartseq2, or M3Drop_UMI. This option specifies the method to be used for identifying variable genes. seurat is the method implemented in the Seurat R package (3). It bins genes according to average expression, then calculates dispersion for each bin as variance to mean ratio. Within each bin, Z-scores are calculated and returned. Z-scores are ranked and the top N are selected. Brennecke2013 refers to the method proposed by Brennecke et al (4). Brennecke2013 estimates and fits technical noise using RNA spikes (technical genes) by fitting a generalized linear model with a gamma function and identity link and the parameterization w=a_1+u+a0. It then uses a chi2 distribution to test the null hypothesis that the squared coefficient of variation does not exceed a certain minimum. FDR<0.10 is considered significant. Currently, Brennecke2013 uses all the genes to estimate noise. scran fits a polynomial regression model to technical noise by modeling the variance versus mean gene expression relationship of ERCC spikes (the original method used local regression) (5). It then decomposes the variance of the biological gene by subtracting the technical variance component and returning the biological variance component. Chen2016 (6) uses linear regression, subsampling, polynomial fitting and gaussian maximum likelihood estimates to derive a set of HVG. M3Drop_smartseq2 models the dropout rate and mean expression using the Michaelis-Menten equation to identify HVG (7). M3Drop_smartseq2 works well with SMART-seq2 data but not UMI data, the former often being sequenced to saturation so zeros are more likely to be dropouts rather than unsaturated sequencing. M3Drop_UMI is the corresponding M3Drop method for UMI data. Default: seurat |
--pca [irlb|regular] |
The PCA method to use. Does not have a big impact on the results. The number of components to use is specified with the --pca_n flag (default is the first 75). |
--hvg_n [int] |
Number of highly variable genes to use. If method is brennecke then --hvg_n determines how many genes will be used from the genes that are significant. Default: 1000 |
--qc_auto [True|False] |
Automatically filters low quality cells using five quality metrics and Mahalanobis distances. Three standard deviations from the mean is considered an outlier and will be removed. Default: True |
--embedding [tSNE|UMAP] |
The method used to project the data to a 2d space. Only used for visualization purposes. t-SNE is more commonly used in scRNA-seq analysis. UMAP may be better at preserving the global structure of the data. Default: tSNE |
--seed [int] |
Set a seed for the random number generator. This setting is used to generate plots and results that are numerically identical. Algorithms such as t-SNE and Fast Truncated Singular Value Decomposition need random numbers. Setting a seed guarantees that the random numbers are the same across sessions. |
--overlay_genes [TEXT] |
Can be used to specify one or more genes for which gene expression will be overlaid on the 2d embedding. The option is useful for examining the expression of individual genes in relation to clusters and cell types. Multiple genes can be given by separating them with comma. If multiple genes are specified, one plot will be generated for each gene. |
--highlight_specific_cells [TEXT] |
Sometimes it can be useful to highlight where a specific cell is falling on the 2d embedding. This option is used to highlight such cells in the scatter plot. Cell identifiers refer to those present in the header of the data matrix. Multiple cell identifiers can be entered separated by commas. |
--violin_top [int] |
Generates violin plots for the top genes of every cluster. The argument specifies how many of the top expressed genes of every cluster are included. "Top" is defined by ranking on the mean within every cluster. |
--timestamp |
Adds a small timestamp to the bottom left corner of every plot. Can be useful when sharing plots in order to distinguish different versions. |
--exclude_gene [TEXT] |
Sometimes we want to exclude certain genes from the analysis. For example tRNA genes or rRNA. This flag can be used to specify a regular expression pattern, which will be matched to the input data and the corresponding genes excluded. |
--annotations [PATH] |
Use this flag to specify a file containing gene annotations. The file should contain two tab-separated columns: one for the genes and one for the annotations. Gene annotation will be added as an additional column in the differential expression analysis files. This option can be useful in case the genome is using systematic gene identifers and not gene symbols. |
--de_direction [any|up|down] |
Specifies the direction of the differential gene expression analysis. Default is up, because usually we want to find genes that are more expressed in one cluster compared to the other. |
A common goal is to define genes that are differentially expressed between cell clusters. alona implements linear models for DE discovery similar to the R package limma. DE analysis is performed by default and the results are written to two files. A linear model y~x is fitted between gene expression and clusters and t statistics and p-values are calculated for coefficients. P-values are two-sided if direction is set to any (otherwise one-sided). The final output for the DE analysis is written into three tables:
This file contains p-values in a matrix format. The number of rows is equal to the number of input genes. Number of columns is equal to the number of comparisons. The column header contains the performed comparisons, e.g. 1_vs_0 indicates that cluster 1 is compared to cluster 0.
This is in long format, separated by tabs, and is generated for easy filtering based on selected cutoffs. Every row corresponds to one hypothesis test. Columns correspond to:
| Column | What it is |
|---|---|
| comparison_A_vs_B | The comparison that was performed. |
| gene | Gene that was tested. |
| p_val | P-value of the test from the t statistic. |
| FDR | False Discovery Rate based on the Benjamini-Hochberg procedure. |
| t_stat | t statistic |
| logFC | log fold change |
| mean.A | mean expression of the gene in cluster A |
| mean.B | mean expression of the gene in cluster B |
| annotations | this column is only added if the --annotation flag is specified |
Contains marker genes by combining p-values of the pairwise tests. P-values are combined with Simes' method.
Please file a bug report through Github.
Limited support is available through e-mail:
- Oscar Franzen [email protected]
A manuscript has been submitted.
GPLv3





















