tidyNano is an R package that imports raw NanoSight data and provides a framework to clean, analyze, and visualize nanoparticle analysis data. You can browse the source code on GitHub.
Check out our manuscript in PLOS ONE!

A representative video of NTA data of murine plasma exosomes from our paper.

Added more reprexes in the README to assist in importing data.
Fixed custom_name argument in nanoimport() function and
nanocombine() function.
nanoimport() has just been updated to be more flexible and works with
NTA versions 3.0 - 3.4! The function should be able to detect and
import your data.
nanolyze() and nanocount() both will ungroup data.
Nomenclature
We recommend naming your files in snake_case so it’s conducive for
processing with the nanotidy() function. With each sample we recommend
adding important information so you can parse it out during analysis, be
sure to influde the dilution value in the sample name if you do not
enter it during the dilution entry box during acquisition. For example
you can name your files using the general formula
SampleID_Treatment_1500_. Be sure to leave a _ at the end so the NTA
software will append your files with a numerical count (00,01,02…) which
will serve as way to track your technical replicates.
If you already collected you data and don’t have your dilution in your
sample name you can simply add the dilution when you use nanoimport().
Simply add the dilution value in the custom_name argument. If you did
label your samples and enter in the dilution in the NTA software during
acquisition you can make auto_name == TRUE in
nanoimport().
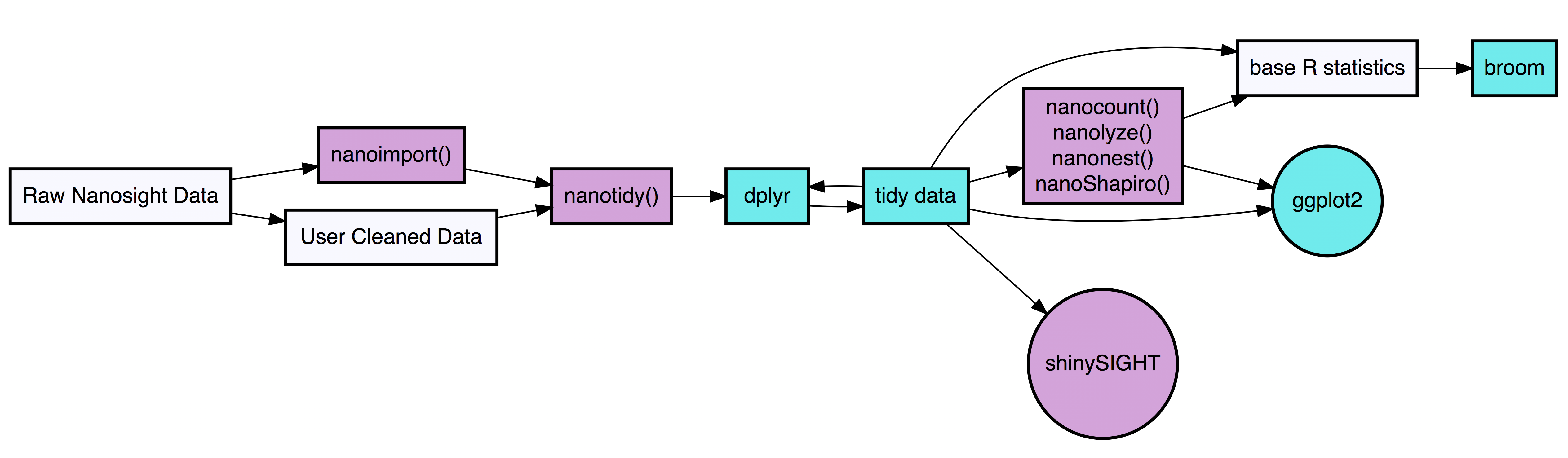
tidyNano functions (Purple) allow for easily extracting and converting raw NTA count data into a tidy dataframe that is suitable for analysis using dplyr and ggplot2. tidyNano also provides a interactive shiny application (shinySIGHT) for visualizing data.
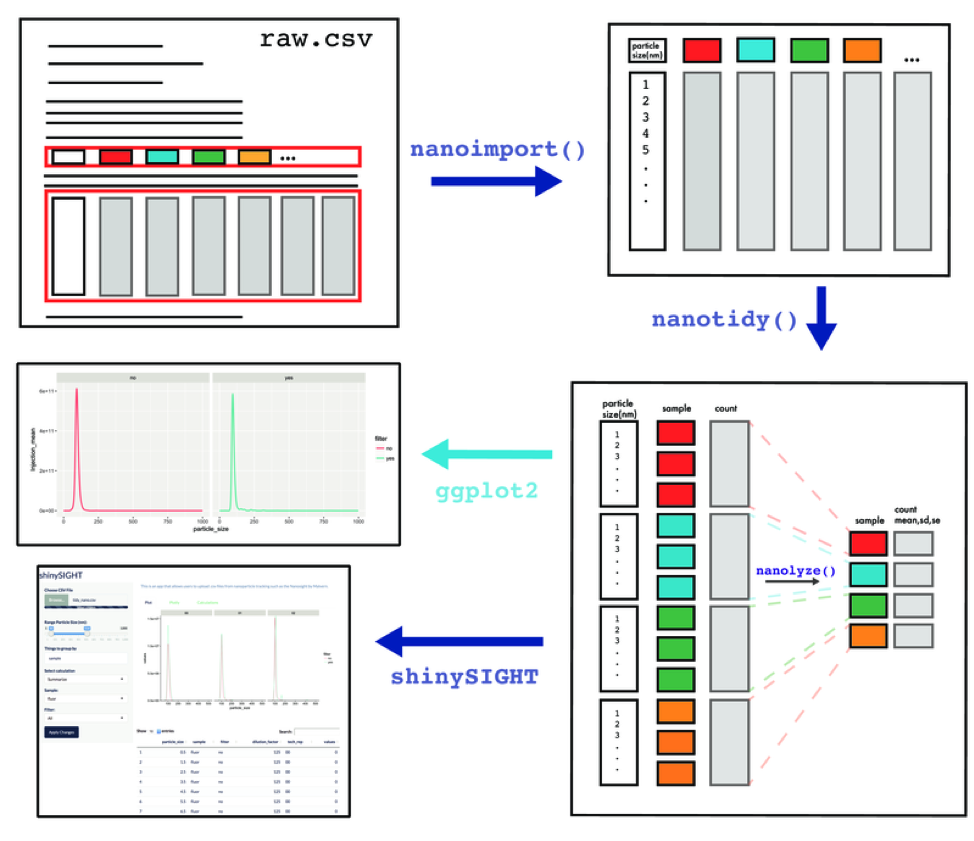
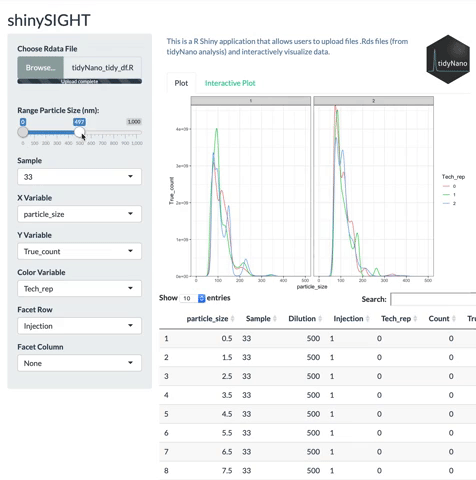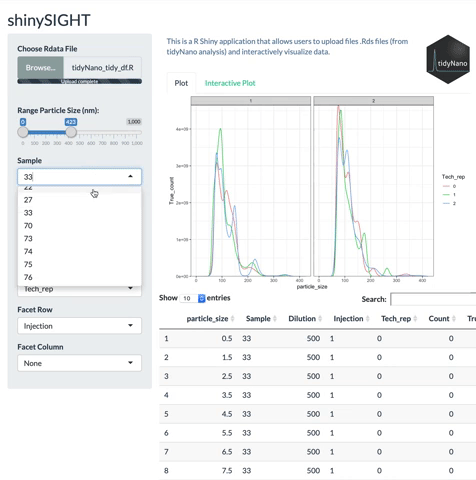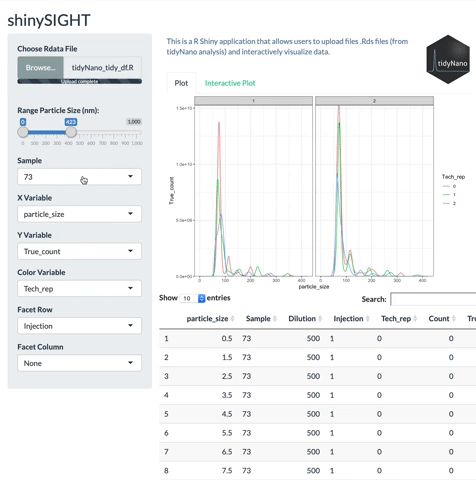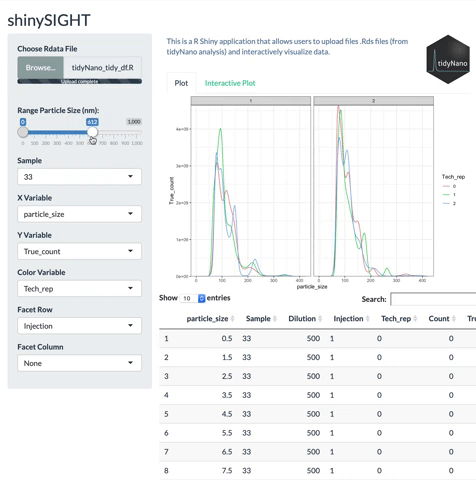
tidyNano also includes a Shiny app named shinySIGHT for interactive visualization of NTA data.
The latest development version can be installed from github:
# install.packages("devtools")
# devtools::install_github("nguyens7/tidyNano")library(tidyNano)
library(tidyverse)
file <- system.file("extdata", "beads.csv", package = "tidyNano")
data <- nanoimport(file)
head(data)
#> particle_size std_10000_yes_2_00 std_10000_yes_2_01 std_10000_yes_2_02
#> 1 0.5 0 0 0
#> 2 1.5 0 0 0
#> 3 2.5 0 0 0
#> 4 3.5 0 0 0
#> 5 4.5 0 0 0
#> 6 5.5 0 0 0
#> std_10000_no_2_00 std_10000_no_2_01 std_10000_no_2_02 std_10000_yes_1_00
#> 1 0 0 0 0
#> 2 0 0 0 0
#> 3 0 0 0 0
#> 4 0 0 0 0
#> 5 0 0 0 0
#> 6 0 0 0 0
#> std_10000_yes_1_01 std_10000_yes_1_02 std_10000_no_1_00 std_10000_no_1_01
#> 1 0 0 0 0
#> 2 0 0 0 0
#> 3 0 0 0 0
#> 4 0 0 0 0
#> 5 0 0 0 0
#> 6 0 0 0 0
#> std_10000_no_1_02
#> 1 0
#> 2 0
#> 3 0
#> 4 0
#> 5 0
#> 6 0nanoimport is a function that extracts the particle data from raw a
nanosight .csv file and creates a dataframe that is suitable for
cleaning within R. Note: This assumes you added the dilution factor
when you named your samples during acquistion.
data <- nanoimport(file)
#> NTA version: 3.2
#> Sample name:
#> Dilution line no: 42
#> Warning in function_list[[k]](value): NAs introduced by coercion
#> Dilution factor detected: NA
#> Auto name = FALSE
#> Custom name: NULL
#> Dilution value: 1 (Didn't parse)
head(data)
#> particle_size std_10000_yes_2_00 std_10000_yes_2_01 std_10000_yes_2_02
#> 1 0.5 0 0 0
#> 2 1.5 0 0 0
#> 3 2.5 0 0 0
#> 4 3.5 0 0 0
#> 5 4.5 0 0 0
#> 6 5.5 0 0 0
#> std_10000_no_2_00 std_10000_no_2_01 std_10000_no_2_02 std_10000_yes_1_00
#> 1 0 0 0 0
#> 2 0 0 0 0
#> 3 0 0 0 0
#> 4 0 0 0 0
#> 5 0 0 0 0
#> 6 0 0 0 0
#> std_10000_yes_1_01 std_10000_yes_1_02 std_10000_no_1_00 std_10000_no_1_01
#> 1 0 0 0 0
#> 2 0 0 0 0
#> 3 0 0 0 0
#> 4 0 0 0 0
#> 5 0 0 0 0
#> 6 0 0 0 0
#> std_10000_no_1_02
#> 1 0
#> 2 0
#> 3 0
#> 4 0
#> 5 0
#> 6 0If you didn’t include your dilution factor in the sample name you can
use the argument auto_name == TRUE within the nanoimport() function.
file2 <- system.file("extdata", "beads2.csv", package = "tidyNano")
data2 <- nanoimport(file2, auto_name = TRUE)
#> NTA version: 3.4
#> Sample name:
#> Dilution line no: 44
#> Dilution factor detected: 10000
#> Auto name: _10000
#> Custom name: NULL
#> Dilution value: 10000
head(data2)
#> particle_size stdv34_00_10000 stdv34_01_10000 stdv34_02_10000 stdv34_03_10000
#> 1 0.5 0 0 0 0
#> 2 1.5 0 0 0 0
#> 3 2.5 0 0 0 0
#> 4 3.5 0 0 0 0
#> 5 4.5 0 0 0 0
#> 6 5.5 0 0 0 0
#> stdv34_04_10000
#> 1 0
#> 2 0
#> 3 0
#> 4 0
#> 5 0
#> 6 0You can even add a custom name to append extra information to your
sample columns using the custom_name()
argument.
custom_name_data2 <- nanoimport(file2, auto_name = TRUE, custom_name = "YourLabelHere")
#> NTA version: 3.4
#> Sample name:
#> Dilution line no: 44
#> Dilution factor detected: 10000
#> Auto name: _YourLabelHere_10000
#> Custom name: YourLabelHere
#> Dilution value: 10000
head(custom_name_data2)
#> particle_size stdv34_00_YourLabelHere_10000 stdv34_01_YourLabelHere_10000
#> 1 0.5 0 0
#> 2 1.5 0 0
#> 3 2.5 0 0
#> 4 3.5 0 0
#> 5 4.5 0 0
#> 6 5.5 0 0
#> stdv34_02_YourLabelHere_10000 stdv34_03_YourLabelHere_10000
#> 1 0 0
#> 2 0 0
#> 3 0 0
#> 4 0 0
#> 5 0 0
#> 6 0 0
#> stdv34_04_YourLabelHere_10000
#> 1 0
#> 2 0
#> 3 0
#> 4 0
#> 5 0
#> 6 0tidy_data <- data %>%
nanotidy(sep_var = c("Sample", "Dilution","Filter","Injection","Tech_rep"))
head(tidy_data)
#> particle_size Sample Dilution Filter Injection Tech_rep Count True_count
#> 1 0.5 std 10000 yes 2 0 0 0
#> 2 1.5 std 10000 yes 2 0 0 0
#> 3 2.5 std 10000 yes 2 0 0 0
#> 4 3.5 std 10000 yes 2 0 0 0
#> 5 4.5 std 10000 yes 2 0 0 0
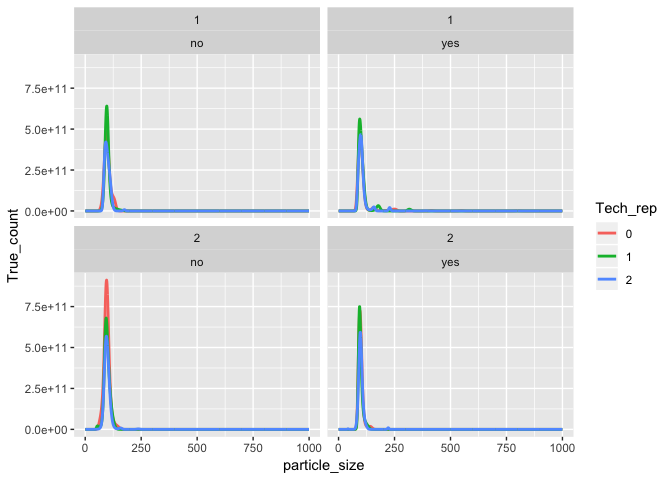
#> 6 5.5 std 10000 yes 2 0 0 0tidy_data %>%
ggplot(aes(x = particle_size, y = True_count, color = Tech_rep)) +
geom_line(size = 1) +
facet_wrap(Injection ~ Filter)