
Welcome to the spatialDLPFC project! This project involves 3 data
types as well as several interactive websites, all of which you are
publicly accessible for you to browse and download.
In this project we studied spatially resolved and single nucleus transcriptomics data from the dorsolateral prefrontal cortex (DLPFC) from postmortem human brain samples. From 10 neurotypical controls we generated spatially-resolved transcriptomics data using using 10x Genomics Visium across the anterior, middle, and posterior DLPFC (n = 30). We also generated single nucleus RNA-seq (snRNA-seq) data using 10x Genomics Chromium from 19 of these tissue blocks. We further generated data from 4 adjacent tissue slices with 10x Genomics Visium Spatial Proteogenomics (SPG), that can be used to benchmark spot deconvolution algorithms. This work is being was performed by the Keri Martinowich, Leonardo Collado-Torres, and Kristen Maynard teams at the Lieber Institute for Brain Development as well as Stephanie Hicks’s group from JHBSPH’s Biostatistics Department.
This project involves the GitHub repositories LieberInstitute/spatialDLPFC and LieberInstitute/DLPFC_snRNAseq.
If you tweet about this website, the data or the R package please use
the #spatialDLPFC hashtag. You can find previous tweets
that way as shown
here.
Thank you for your interest in our work!
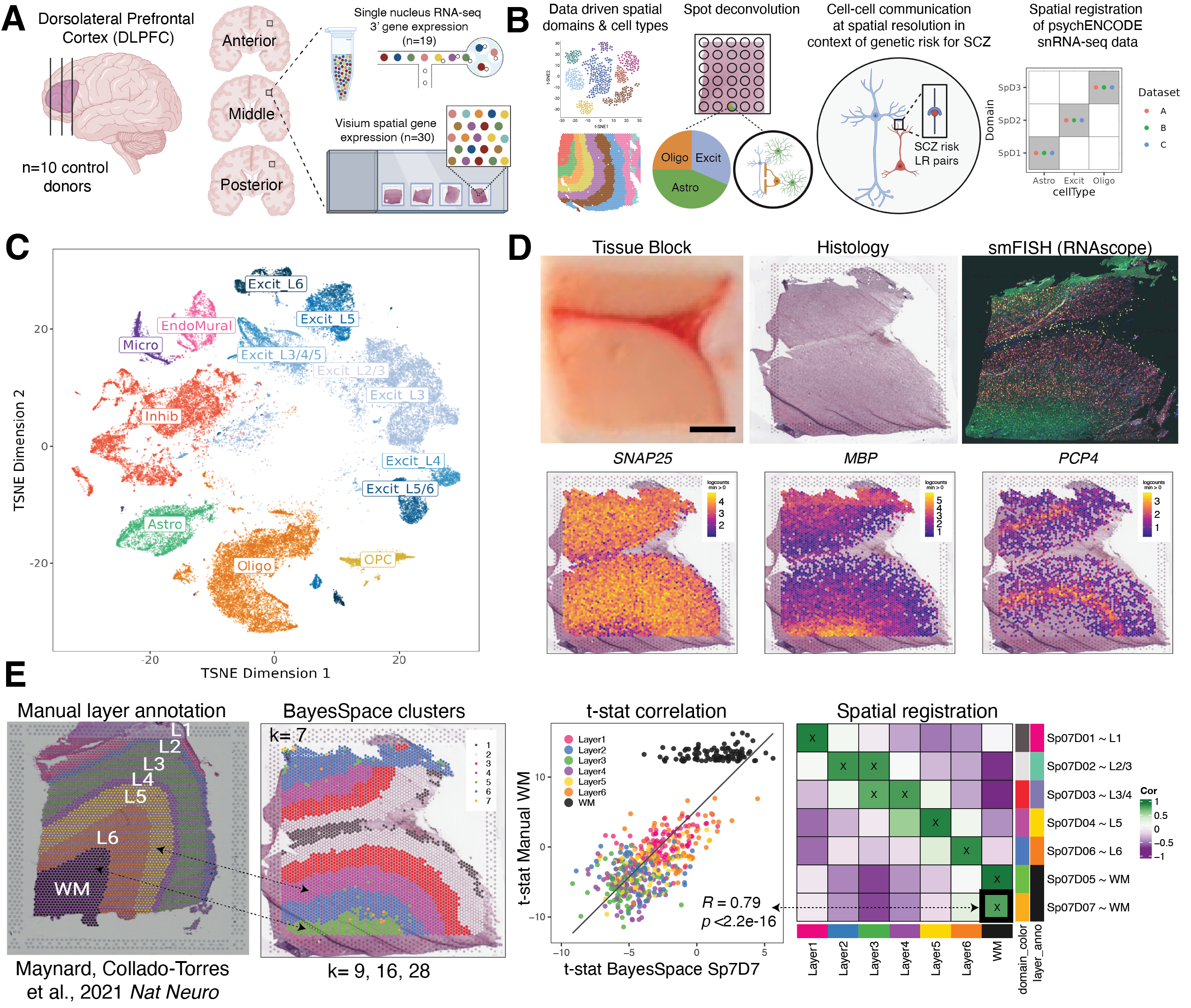
Study design to generate paired single nucleus RNA-sequencing (snRNA-seq) and spatially-resolved transcriptomic data across DLPFC. (A) DLPFC tissue blocks were dissected across the rostral-caudal axis from 10 adult neurotypical control postmortem human brains, including anterior (Ant), middle (Mid), and posterior (Post) positions (n=3 blocks per donor, n=30 blocks total). The same tissue blocks were used for snRNA-seq (10x Genomics 3’ gene expression assay, n=1-2 blocks per donor, n=19 samples) and spatial transcriptomics (10x Genomics Visium spatial gene expression assay, n=3 blocks per donor, n=30 samples). (B) Paired snRNA-seq and Visium data were used to identify data-driven spatial domains (SpDs) and cell types, perform spot deconvolution, conduct cell-cell communication analyses, and spatially register companion PsychENCODE snRNA-seq DLPFC data. (C) t-distributed stochastic neighbor embedding (t-SNE) summarizing layer resolution cell types identified by snRNA-seq. (D) Tissue block orientation and morphology was confirmed by hematoxylin and eosin (H&E) staining and single molecule fluorescent in situ hybridization (smFISH) with RNAscope (SLC17A7 marking excitatory neurons in pink, MBP marking white matter (WM) in green, RELN marking layer (L)1 in yellow, and NR4A2 marking L6 in orange). Scale bar is 2mm. Spotplots depicting log transformed normalized expression (logcounts) of SNAP25, MBP, and PCP4 in the Visium data confirm the presence of gray matter, WM, and cortical layers, respectively. (E) Schematic of unsupervised SpD identification and registration using BayesSpace SpDs at k=7. Enrichment t-statistics computed on BayesSpace SpDs were correlated with manual histological layer annotations from (Maynard, Collado-Torres et al., 2021, Nat Neuro) to map SpDs to known histological layers. The heatmap of correlation values summarizes the relationship between BayesSpace SpDs and classic histological layers. Higher confidence annotations (⍴ > 0.25, merge ratio = 0.1) are marked with an “X”.
All of these interactive websites are powered by open source software, namely:
- 🔭
spatialLIBD - 🔍
samui - 👀
iSEE
We provide the following interactive websites, organized by dataset with software labeled by emojis:
- Visium (n = 30)
- 🔭
spatialDLPFC_Visium_Sp09:
spatialLIBDwebsite showing the spatially-resolved Visium data (n = 30) with statistical results comparing the Sp09 domains. - 🔭 spatialDLPFC_Visium_Sp16: similar but with the Sp16 domains.
- 🔭 spatialDLPFC_Visium_Sp09_position: similar to spatialDLPFC_Visium_Sp09 but with statistical results across the position (anterior, middle, posterior) adjusting for the Sp09 domains.
- 🔭
spatialDLPFC_Visium_Sp09_position_noWM:
similar to spatialDLPFC_Visium_Sp09_position but after dropping
the
SP28D06,SP28D16,SP28D17,SP28D20andSP28D28spots which correspond to white matter (hence thenoWMacronym). - 👀
spatialDLPFC_Visium_Sp09_pseudobulk:
iSEEwebsite showing the pseudo-bulked Sp09 domains spatial data. - 👀 spatialDLPFC_Visium_Sp16_pseudobulk: similar to spatialDLPFC_Visium_Sp09_pseudobulk but with the Sp16 domains data.
- 👀 spatialDLPFC_Visium_Sp28_pseudobulk: similar to spatialDLPFC_Visium_Sp09_pseudobulk but with the Sp28 domains data.
- 🔍 spatialDLPFC Visium on
Samui:
samuiwebsite that allows to zoom in at the spot or cell level.
- 🔭
spatialDLPFC_Visium_Sp09:
- snRNA-seq (n = 19)
- 👀
spatialDLPFC_snRNA-seq:
iSEEwebsite showing the n = 19 snRNA-seq samples at single nucleus resolution.
- 👀
spatialDLPFC_snRNA-seq:
- Visium SPG (n = 4)
- 🔭
spatialDLPFC_Visium_SPG:
spatialLIBDwebsite showing the spatially-resolved data Visium SPG (n = 4). - 🔍 spatialDLPFC Visium SPG on
Samui:
samuiwebsite that allows to zoom in at the spot or cell level.
- 🔭
spatialDLPFC_Visium_SPG:
If you are interested in running the
spatialLIBD applications
locally, you can do so thanks to the
spatialLIBD::run_app(),
which you can also use with your own data as shown in our vignette for
publicly available datasets provided by 10x
Genomics.
## Run this web application locally with:
spatialLIBD::run_app()
## You will have more control about the length of the session and memory usage.
## See http://research.libd.org/spatialLIBD/reference/run_app.html#examples
## for the full R code to run https://libd.shinyapps.io/spatialDLPFC_Visium_Sp09
## locally. See also:
## * https://github.com/LieberInstitute/spatialDLPFC/tree/main/code/deploy_app_k09
## * https://github.com/LieberInstitute/spatialDLPFC/tree/main/code/deploy_app_k09_position
## * https://github.com/LieberInstitute/spatialDLPFC/tree/main/code/deploy_app_k09_position_noWM
## * https://github.com/LieberInstitute/spatialDLPFC/tree/main/code/deploy_app_k16
## * https://github.com/LieberInstitute/spatialDLPFC/tree/main/code/analysis_IF/03_spatialLIBD_app
## You could also use spatialLIBD::run_app() to visualize your
## own data given some requirements described
## in detail in the package vignette documentation
## at http://research.libd.org/spatialLIBD/.We value public questions, as they allow other users to learn from the answers. If you have any questions, please ask them at LieberInstitute/spatialDLPFC/issues and refrain from emailing us. Thank you again for your interest in our work!
Please cite this manuscript if you use data from this project.
Integrated single cell and unsupervised spatial transcriptomic analysis defines molecular anatomy of the human dorsolateral prefrontal cortex Louise A. Huuki-Myers, Abby Spangler, Nicholas J. Eagles, Kelsey D. Montgomery, Sang Ho Kwon, Boyi Guo, Melissa Grant-Peters, Heena R. Divecha, Madhavi Tippani, Chaichontat Sriworarat, Annie B. Nguyen, Prashanthi Ravichandran, Matthew N. Tran, Arta Seyedian, PsychENCODE Consortium, Thomas M. Hyde, Joel E. Kleinman, Alexis Battle, Stephanie C. Page, Mina Ryten, Stephanie C. Hicks, Keri Martinowich, Leonardo Collado-Torres, Kristen R. Maynard bioRxiv 2023.02.15.528722; doi: https://doi.org/10.1101/2023.02.15.528722
Below is the citation in BibTeX format.
@article {Huuki-Myers2023.02.15.528722,
author = {Huuki-Myers, Louise A. and Spangler, Abby and Eagles, Nicholas J. and Montgomery, Kelsey D. and Kwon, Sang Ho and Guo, Boyi and Grant-Peters, Melissa and Divecha, Heena R. and Tippani, Madhavi and Sriworarat, Chaichontat and Nguyen, Annie B. and Ravichandran, Prashanthi and Tran, Matthew N. and Seyedian, Arta and , and Hyde, Thomas M. and Kleinman, Joel E. and Battle, Alexis and Page, Stephanie C. and Ryten, Mina and Hicks, Stephanie C. and Martinowich, Keri and Collado-Torres, Leonardo and Maynard, Kristen R.},
title = {Integrated single cell and unsupervised spatial transcriptomic analysis defines molecular anatomy of the human dorsolateral prefrontal cortex},
elocation-id = {2023.02.15.528722},
year = {2023},
doi = {10.1101/2023.02.15.528722},
publisher = {Cold Spring Harbor Laboratory},
URL = {https://www.biorxiv.org/content/early/2023/02/15/2023.02.15.528722},
eprint = {https://www.biorxiv.org/content/early/2023/02/15/2023.02.15.528722.full.pdf},
journal = {bioRxiv}
}
Below is the citation output from using citation('spatialLIBD') in R.
Please run this yourself to check for any updates on how to cite
spatialLIBD.
print(citation("spatialLIBD")[1], bibtex = TRUE)
#> Pardo B, Spangler A, Weber LM, Hicks SC, Jaffe AE, Martinowich K,
#> Maynard KR, Collado-Torres L (2022). "spatialLIBD: an R/Bioconductor
#> package to visualize spatially-resolved transcriptomics data." _BMC
#> Genomics_. doi:10.1186/s12864-022-08601-w
#> <https://doi.org/10.1186/s12864-022-08601-w>,
#> <https://doi.org/10.1186/s12864-022-08601-w>.
#>
#> A BibTeX entry for LaTeX users is
#>
#> @Article{,
#> title = {spatialLIBD: an R/Bioconductor package to visualize spatially-resolved transcriptomics data},
#> author = {Brenda Pardo and Abby Spangler and Lukas M. Weber and Stephanie C. Hicks and Andrew E. Jaffe and Keri Martinowich and Kristen R. Maynard and Leonardo Collado-Torres},
#> year = {2022},
#> journal = {BMC Genomics},
#> doi = {10.1186/s12864-022-08601-w},
#> url = {https://doi.org/10.1186/s12864-022-08601-w},
#> }Please note that the spatialLIBD was only made possible thanks to many
other R and bioinformatics software authors, which are cited either in
the vignettes and/or the paper(s) describing the package.
To cite samui please use:
Performant web-based interactive visualization tool for spatially-resolved transcriptomics experiments Chaichontat Sriworarat, Annie Nguyen, Nicholas J. Eagles, Leonardo Collado-Torres, Keri Martinowich, Kristen R. Maynard, Stephanie C. Hicks Biological Imaging; doi: https://doi.org/10.1017/S2633903X2300017X
Below is the citation in BibTeX format.
@article{sriworarat_performant_2023,
title = {Performant web-based interactive visualization tool for spatially-resolved transcriptomics experiments},
volume = {3},
issn = {2633-903X},
url = {https://www.cambridge.org/core/journals/biological-imaging/article/performant-webbased-interactive-visualization-tool-for-spatiallyresolved-transcriptomics-experiments/B66303984D10B9E5A23D3656CB8537C0},
doi = {10.1017/S2633903X2300017X},
language = {en},
urldate = {2024-04-19},
journal = {Biological Imaging},
author = {Sriworarat, Chaichontat and Nguyen, Annie and Eagles, Nicholas J. and Collado-Torres, Leonardo and Martinowich, Keri and Maynard, Kristen R. and Hicks, Stephanie C.},
month = jan,
year = {2023},
keywords = {georeferencing, interactive image viewer, multi-dimensional image, single-cell transcriptomics, spatially resolved transcriptomics, web-based browser},
pages = {e15}
}
To cite VistoSeg please use:
VistoSeg: {Processing utilities for high-resolution images for spatially resolved transcriptomics data. Madhavi Tippani, Heena R. Divecha, Joseph L. Catallini II, Sang Ho Kwon, Lukas M. Weber, Abby Spangler, Andrew E. Jaffe, Thomas M. Hyde, Joel E. Kleinman, Stephanie C. Hicks, Keri Martinowich, Leonardo Collado-Torres, Stephanie C. Page, Kristen R. Maynard Biological Imaging ; doi: https://doi.org/10.1017/S2633903X23000235
Below is the citation in BibTeX format.
@article{tippani_vistoseg_2023,
title = {{VistoSeg}: {Processing} utilities for high-resolution images for spatially resolved transcriptomics data},
volume = {3},
issn = {2633-903X},
shorttitle = {{VistoSeg}},
url = {https://www.cambridge.org/core/journals/biological-imaging/article/vistoseg-processing-utilities-for-highresolution-images-for-spatially-resolved-transcriptomics-data/990CBC4AC069F5EDC62316919398404B},
doi = {10.1017/S2633903X23000235},
language = {en},
urldate = {2024-04-19},
journal = {Biological Imaging},
author = {Tippani, Madhavi and Divecha, Heena R. and Catallini, Joseph L. and Kwon, Sang H. and Weber, Lukas M. and Spangler, Abby and Jaffe, Andrew E. and Hyde, Thomas M. and Kleinman, Joel E. and Hicks, Stephanie C. and Martinowich, Keri and Collado-Torres, Leonardo and Page, Stephanie C. and Maynard, Kristen R.},
month = jan,
year = {2023},
keywords = {hematoxylin and eosin, immunofluorescence, MATLAB, segmentation, spatially resolved transcriptomics, Visium, Visium-Spatial Proteogenomics},
pages = {e23}
}
We highly value open data sharing and believe that doing so accelerates
science, as was the case between our
HumanPilot and the
external BayesSpace
projects, documented on this
slide.
spatialLIBD also allows
you to access the data from this project as ready to use R objects. That
is, a:
SpatialExperimentobject for the Visium samples (n = 30)SpatialExperimentobject for the Visium SPG samples (n = 4)SingleCellExperimentobject for the snRNA-seq samples (n = 19)
You can use the
zellkonverter
Bioconductor package to convert any of them into Python
AnnData objects. If you
browse our code, you can find examples of such conversions.
If you are unfamiliar with these tools, you might want to check the LIBD rstats club (check and search keywords on the schedule) videos and resources.
Get the latest stable R release from
CRAN. Then install spatialLIBD from
Bioconductor with the following code:
## Install BiocManager in order to install Bioconductor packages properly
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
## Check that you have a valid R/Bioconductor installation
BiocManager::valid()
## Now install spatialLIBD from Bioconductor
## (this version has been tested on macOS, winOS, linux)
BiocManager::install("spatialLIBD")
## If you need the development version from GitHub you can use the following:
# BiocManager::install("LieberInstitute/spatialLIBD")
## Note that this version might include changes that have not been tested
## properly on all operating systems.Using spatialLIBD you can access the spatialDLPFC transcriptomics data
from the 10x Genomics Visium platform. For example, this is the code you
can use to access the spatially-resolved data. For more details, check
the help file for fetch_data().
## Check that you have a recent version of spatialLIBD installed
stopifnot(packageVersion("spatialLIBD") >= "1.11.6")
## Download the spot-level data
spe <- spatialLIBD::fetch_data(type = "spatialDLPFC_Visium")
## This is a SpatialExperiment object
spe
#> class: SpatialExperiment
#> dim: 28916 113927
#> metadata(1): BayesSpace.data
#> assays(2): counts logcounts
#> rownames(28916): ENSG00000243485 ENSG00000238009 ... ENSG00000278817 ENSG00000277196
#> rowData names(7): source type ... gene_type gene_search
#> colnames(113927): AAACAACGAATAGTTC-1 AAACAAGTATCTCCCA-1 ... TTGTTTGTATTACACG-1 TTGTTTGTGTAAATTC-1
#> colData names(155): age array_col ... VistoSeg_proportion wrinkle_type
#> reducedDimNames(8): 10x_pca 10x_tsne ... HARMONY UMAP.HARMONY
#> mainExpName: NULL
#> altExpNames(0):
#> spatialCoords names(2) : pxl_col_in_fullres pxl_row_in_fullres
#> imgData names(4): sample_id image_id data scaleFactor
## Note the memory size
lobstr::obj_size(spe)
#> 6.97 GB
## Set the cluster colors
colors_BayesSpace <- Polychrome::palette36.colors(28)
names(colors_BayesSpace) <- seq_len(28)
## Remake the logo image with histology information
p09 <- spatialLIBD::vis_clus(
spe = spe,
clustervar = "BayesSpace_harmony_09",
sampleid = "Br6522_ant",
colors = colors_BayesSpace,
... = " spatialDLPFC Human Brain\nSp09 domains -- made with spatialLIBD"
)
p09## Repeat but for Sp16
p16 <- spatialLIBD::vis_clus(
spe = spe,
clustervar = "BayesSpace_harmony_16",
sampleid = "Br6522_ant",
colors = colors_BayesSpace,
... = " spatialDLPFC Human Brain\nSp16 domains -- made with spatialLIBD"
)
p16
The source data described in this manuscript are available via the PsychENCODE Knowledge Portal (https://PsychENCODE.synapse.org/). The PsychENCODE Knowledge Portal is a platform for accessing data, analyses, and tools generated through grants funded by the National Institute of Mental Health (NIMH) PsychENCODE Consortium. Data is available for general research use according to the following requirements for data access and data attribution: (https://PsychENCODE.synapse.org/DataAccess). For access to content described in this manuscript see: https://doi.org/10.7303/syn51032055.1 or https://www.synapse.org/#!Synapse:syn51032055/datasets/. All figure and table files are available from https://www.synapse.org/#!Synapse:syn50908929.
You can also access all the raw data through
Globus (jhpce#spatialDLPFC and
jhpce#DLPFC_snRNAseq). This includes all the input FASTQ files as well
as the outputs from tools such as
SpaceRanger
or
CellRanger.
The files are mostly organized following the
LieberInstitute/template_project
project structure.
- JHPCE locations:
/dcs04/lieber/lcolladotor/spatialDLPFC_LIBD4035/spatialDLPFC/dcs04/lieber/lcolladotor/deconvolution_LIBD4030/DLPFC_snRNAseq
- Slack channel:
libd_dlpfc_spatial.
Files: spatialDLPFC
code: R, python, and shell scripts for running various analyses.spot_deconvo: cell-type deconvolution within Visium spots, enabled by tools liketangram,cell2location,cellpose, andSPOTlightspython: older legacy testing scripts mostly replaced byspot_deconvo
plots: plots generated by RMarkdown or R analysis scripts in.pdfor.pngformatprocessed-dataimages_spatialLIBD: images used for runningSpaceRangerNextSeq:SpaceRangeroutput filesrdata: R objects
raw-dataFASTQ: FASTQ files fromNextSeqruns.FASTQ_renamed: renamed symbolic links to the original FASTQs, with consistent nomenclatureImages: raw images from the scanner in.tifformat and around 3 GB per sample.images_raw_align_jsonpsychENCODE: external data from PsychENCODE (doi: 10.7303/syn2787333).sample_info: spreadsheet with information about samples (sample ID, sample name, slide serial number, capture area ID)
This GitHub repository is organized along the R/Bioconductor-powered
Team Data Science group
guidelines.
It aims to follow the
LieberInstitute/template_project
structure, though most of the code/analysis output is saved at
processed-data/rdata/spe directory unlike what’s specified in the
template structure. This is due to historical reasons.
Files: DLPFC_snRNAseq
code: R scripts for running various analyses.plots: plots generated by RMarkdown or R analysis scripts in.pdfor.pngformatprocessed-datacellranger:CellRangeroutput files
raw-dataFASTQ: FASTQ files.sample_info: spreadsheet with information about samples (sample ID, sample name)
This GitHub repository is organized along the R/Bioconductor-powered Team Data Science group guidelines. It aims to follow the LieberInstitute/template_project structure.
- Reference transcriptome from 10x Genomics:
/dcs04/lieber/lcolladotor/annotationFiles_LIBD001/10x/refdata-gex-GRCh38-2020-A/


















