haessar / peaks2utr Goto Github PK
View Code? Open in Web Editor NEWA robust Python tool for the annotation of 3’ UTRs
Home Page: https://doi.org/10.1093/bioinformatics/btad112
License: GNU General Public License v3.0
A robust Python tool for the annotation of 3’ UTRs
Home Page: https://doi.org/10.1093/bioinformatics/btad112
License: GNU General Public License v3.0














Hi, thanks for a great software!
I was trying out peaks2utr on my data and got this "too many open files " error.
I'm running it on a institutional cluster server that does not allow changing ulimit -n.
Is there something that I can do to fix this?
INFO Iterating over peaks to annotate 3' UTRs.: 14%|█████▉ | 2291/15932 [01:50<07:16, 31.28it/s]Process Process-3:
Traceback (most recent call last):
File "/lustre/home/abs/anaconda3/envs/peaks2utr/lib/python3.10/multiprocessing/process.py", line 314, in _bootstrap
self.run()
File "/lustre/home/abs/anaconda3/envs/peaks2utr/lib/python3.10/multiprocessing/process.py", line 108, in run
self._target(*self._args, **self._kwargs)
File "/lustre/home/abs/anaconda3/envs/peaks2utr/lib/python3.10/site-packages/peaks2utr/annotations.py", line 52, in _iter_peaks
annotate_utr_for_peak(db, queue, peak, args.max_distance, args.override_utr, args.extend_utr, args.five_prime_ext)
File "/lustre/home/abs/anaconda3/envs/peaks2utr/lib/python3.10/site-packages/peaks2utr/annotations.py", line 82, in annotate_utr_for_peak
bt = pybedtools.BedTool(cached(k + "_coverage_gaps.bed")).filter(lambda x: x.chrom == peak.chr)
File "/lustre/home/abs/anaconda3/envs/peaks2utr/lib/python3.10/site-packages/pybedtools/bedtool.py", line 529, in __init__
self._isbam = isBAM(fn)
File "/lustre/home/abs/anaconda3/envs/peaks2utr/lib/python3.10/site-packages/pybedtools/helpers.py", line 214, in isBAM
if isBGZIP(fn) and (in_.read(4).decode() == "BAM\x01"):
File "/lustre/home/abs/anaconda3/envs/peaks2utr/lib/python3.10/site-packages/pybedtools/helpers.py", line 182, in isBGZIP
with open(fn, "rb") as fh:
OSError: [Errno 24] Too many open files: '/path/to/.cache/forward_coverage_gaps.bed'
Just to be clear, I installed peaks2utr through pip, in a conda environment created as below.
mamba create -n peaks2utr -c bioconda genometools-genometools bedtools python numpy
pip install MACS3
pip install peaks2utr
I installed peaks2utr following the steps I reported in another issue (#1) and tried to execute the example run (using the files in the demo folder).
As I mentioned in #1, my Python version (that has been installed to the virtual environment) is
Python 3.8.15
However, the example run was not successful.
A possible reason could be that it is a newly created virtual environment which may lack some of the basic packages required to run peaks2utr.
Here is what the error messages looked like:
peaks2utr Tb927_01_v5.1.gff Tb927_01_v5.1.slice.bam
2023-01-16 16:13:56,204 - INFO - Make .log directory
2023-01-16 16:13:56,205 - INFO - Make .cache directory
2023-01-16 16:13:56,206 - INFO - Splitting forward strand from Tb927_01_v5.1.slice.bam.
2023-01-16 16:13:58,190 - INFO - Finished splitting forward strand.
2023-01-16 16:13:58,190 - INFO - Splitting reverse strand from Tb927_01_v5.1.slice.bam.
2023-01-16 16:13:58,520 - INFO - Finished splitting reverse strand.
2023-01-16 16:13:58,521 - INFO - Splitting forward-stranded BAM file into read-groups.
2023-01-16 16:14:00,566 - INFO - Splitting reverse-stranded BAM file into read-groups.
2023-01-16 16:14:00,886 - INFO - Indexing ~/demo/.cache/Tb927_01_v5.1.slice.forward_0.bam.
2023-01-16 16:14:00,915 - INFO - Indexing ~/demo/.cache/Tb927_01_v5.1.slice.forward_3.bam.
2023-01-16 16:14:00,943 - INFO - Indexing ~/demo/.cache/Tb927_01_v5.1.slice.forward_2.bam.
2023-01-16 16:14:00,971 - INFO - Indexing ~/demo/.cache/Tb927_01_v5.1.slice.forward_1.bam.
2023-01-16 16:14:01,000 - INFO - Indexing ~/demo/.cache/Tb927_01_v5.1.slice.reverse_1.bam.
2023-01-16 16:14:01,005 - INFO - Indexing ~/demo/.cache/Tb927_01_v5.1.slice.reverse_2.bam.
2023-01-16 16:14:01,010 - INFO - Indexing ~/demo/.cache/Tb927_01_v5.1.slice.reverse_0.bam.
2023-01-16 16:14:01,015 - INFO - Indexing ~/demo/.cache/Tb927_01_v5.1.slice.reverse_3.bam.
INFO Iterating over reads to determine SPAT pileups: 0%| | [00:00<?]
2023-01-16 16:14:01,060 - INFO - Clearing cache.
Traceback (most recent call last):
File "/~~~~home~~~/opt/miniconda3/envs/peaks2utr_v2/bin/peaks2utr", line 8, in <module>
sys.exit(main())
File "/~~~~home~~~/opt/miniconda3/envs/peaks2utr_v2/lib/python3.8/site-packages/peaks2utr/__init__.py", line 49, in main
asyncio.run(_main())
File "/~~~~home~~~/opt/miniconda3/envs/peaks2utr_v2/lib/python3.8/asyncio/runners.py", line 44, in run
return loop.run_until_complete(main)
File "/~~~~home~~~/opt/miniconda3/envs/peaks2utr_v2/lib/python3.8/asyncio/base_events.py", line 616, in run_until_complete
return future.result()
File "/~~~~home~~~/opt/miniconda3/envs/peaks2utr_v2/lib/python3.8/site-packages/peaks2utr/__init__.py", line 100, in _main
bs.pileup_soft_clipped_reads()
File "/~~~~home~~~/opt/miniconda3/envs/peaks2utr_v2/lib/python3.8/site-packages/peaks2utr/preprocess.py", line 85, in pileup_soft_clipped_reads
multiprocess_over_dict(self._count_unmapped_pileups, self.outputs)
File "/~~~~home~~~/opt/miniconda3/envs/peaks2utr_v2/lib/python3.8/site-packages/peaks2utr/utils.py", line 51, in multiprocess_over_dict
p.start()
File "/~~~~home~~~/opt/miniconda3/envs/peaks2utr_v2/lib/python3.8/multiprocessing/process.py", line 121, in start
self._popen = self._Popen(self)
File "/~~~~home~~~/opt/miniconda3/envs/peaks2utr_v2/lib/python3.8/multiprocessing/context.py", line 224, in _Popen
return _default_context.get_context().Process._Popen(process_obj)
File "/~~~~home~~~/opt/miniconda3/envs/peaks2utr_v2/lib/python3.8/multiprocessing/context.py", line 284, in _Popen
return Popen(process_obj)
File "/~~~~home~~~/opt/miniconda3/envs/peaks2utr_v2/lib/python3.8/multiprocessing/popen_spawn_posix.py", line 32, in __init__
super().__init__(process_obj)
File "/~~~~home~~~/opt/miniconda3/envs/peaks2utr_v2/lib/python3.8/multiprocessing/popen_fork.py", line 19, in __init__
self._launch(process_obj)
File "/~~~~home~~~/opt/miniconda3/envs/peaks2utr_v2/lib/python3.8/multiprocessing/popen_spawn_posix.py", line 47, in _launch
reduction.dump(process_obj, fp)
File "/~~~~home~~~/opt/miniconda3/envs/peaks2utr_v2/lib/python3.8/multiprocessing/reduction.py", line 60, in dump
ForkingPickler(file, protocol).dump(obj)
TypeError: cannot pickle '_io.TextIOWrapper' object
where I have manually replaced the path of the home directory of the cloned peaks2utr folder as ~, and where my miniconda has been installed as /~~~~home~~~/.
So, as the error message indicated, it seems something's wrong when the script tried to call some function in the multiprocessing module? I'd be grateful if you could kindly point out how I could potentially solve this. Perhaps some packages are missing in this newly created virtual environment?
Thank you so much.
Hi Haessar,
Every run of peaks2utr I have, including the run from the demo, has these message in the run log:
[E::idx_find_and_load] Could not retrieve index file for '/home/jhc103/code/rosetta-stone-proj/peaks2utr-demo/.cache/Tb927_01_v5.1.slice.forward.bam'
[E::idx_find_and_load] Could not retrieve index file for '/home/jhc103/code/rosetta-stone-proj/peaks2utr-demo/.cache/Tb927_01_v5.1.slice.reverse.bam'
Is this expected behavior? I am running version 0.5.1. I did not switch to the latest peaks2utr because I was not able to get those versions to work on my files.
Best
Hello,
I have run peaks2utr to improve the 3'UTR annotation of the Japanese quail genome, and I always use the -gtf flag to get a GTF file output. When I open the GTF file output as a data frame in Rstudio, I noticed that an extra variable 'colour' has been added, what exactly is this?
Thank you in advance!
Hello,
I was able to successfully install peaks2utr but I tried to execute demo I got this error:
TypeError: cannot pickle '_io.TextIOWrapper' object
How can this be solved?
Thanks.
Hi Haessa,
I am using peak2utr 1.0.0 with gffutils 0.10.1, this is the error I get when I run it on my gff file. Is this an issue with the format of my gff or bam file? (I've attached my full error log as well)
File "/home/jhc103/miniconda3/envs/toolshed-peaks2utr-latest/lib/python3.9/site-packages/gffutils/create.py", line 507, in create
self._populate_from_lines(self.iterator)
File "/home/jhc103/miniconda3/envs/toolshed-peaks2utr-latest/lib/python3.9/site-packages/gffutils/create.py", line 591, in _populate_from_lines
fixed, final_strategy = self._do_merge(f, self.merge_strategy)
File "/home/jhc103/miniconda3/envs/toolshed-peaks2utr-latest/lib/python3.9/site-packages/gffutils/create.py", line 226, in _do_merge
raise ValueError("Duplicate ID {0.id}".format(f))
ValueError: Duplicate ID cds-XP_003638833.2
Thanks!
Seqids of Gtf/Gff and BAM file should match. Check should be performed immediately in workflow.
Would you recommend one to use bam files from bulk RNA-seq alignment to extend 3' UTR. i.e. if one has bulk RNA-seq data from tsetse salivary gland infected with Trypanosoma brucei and single cell RNA-seq of Trypanosoma brucei from tsetse salivary gland, use the bam file of bulk alignment to extend 3' UTR and use the new generated gff/gtf file with utrs for scRNA-seq processing. Can one use the bam files generated by any alignment tool ie. bowtie2, Hisat2, Star etc?
Hi,
First of all thanks for this tool, I work with many non-traditional model animals and this has been great for improving the in-house gene annotations we have for them. peaks2utr has worked for me on 3 different species, but now I keep running into the same error with one specific species (ateAlb), and I don't understand what the issue is.
I have been running peaks2utr as follows:
> peaks2utr --extend-utr -p 10 -o $outGtf --gtf $inGtf $inBam
#inBam = a merge of 10X scRNAseq bam files from ateAlb
#inGtf = in house annotation in gff3 format
> head $inGtf
ateAlb2.1 funannotate transcript 76692 86162 . - . transcript_id "Aalb_00004274-T1"; gene_id "Aalb_00004274"
ateAlb2.1 funannotate exon 76692 79108 . - . transcript_id "Aalb_00004274-T1"; gene_id "Aalb_00004274";
ateAlb2.1 funannotate exon 82165 82457 . - . transcript_id "Aalb_00004274-T1"; gene_id "Aalb_00004274";
ateAlb2.1 funannotate exon 84823 85591 . - . transcript_id "Aalb_00004274-T1"; gene_id "Aalb_00004274";
ateAlb2.1 funannotate exon 85672 85898 . - . transcript_id "Aalb_00004274-T1"; gene_id "Aalb_00004274";
ateAlb2.1 funannotate exon 85993 86162 . - . transcript_id "Aalb_00004274-T1"; gene_id "Aalb_00004274";
ateAlb2.1 funannotate transcript 153660 157786 . + . transcript_id "Aalb_00004275-T1"; gene_id "Aalb_00004275"
ateAlb2.1 funannotate exon 153660 155351 . + . transcript_id "Aalb_00004275-T1"; gene_id "Aalb_00004275";
ateAlb2.1 funannotate exon 155474 156732 . + . transcript_id "Aalb_00004275-T1"; gene_id "Aalb_00004275";
ateAlb2.1 funannotate exon 156956 156983 . + . transcript_id "Aalb_00004275-T1"; gene_id "Aalb_00004275";
I attached is the full terminal output for the peaks2utr run with the whole traceback for the errors, but these are the two main error messages I get:
File "/home/.local/lib/python3.10/site-packages/gffutils/bins.py", line 79, in bins
if start >= MAX_CHROM_SIZE or stop >= MAX_CHROM_SIZE:
TypeError: '>=' not supported between instances of 'NoneType' and 'int'
and
File "/pkg/python-3.10.10-1/lib/python3.10/shutil.py", line 729, in rmtree
os.rmdir(path)
OSError: [Errno 39] Directory not empty: '/path/to/.cache'
Could you help solve this issue? I don't even now where to begin with as the cache should be cleared (it does state 2023-11-03 20:54:57,929 - INFO - Clearing cache. in the log), nor do I understand where the NoneType is coming from. I'm using peaks2utr 1.1.0.
Thank you in advance,
Anna
> ls ./.cache
forward_peaks.broadPeak forward_peaks.xls reverse_peaks.gappedPeak
forward_peaks.gappedPeak reverse_peaks.broadPeak reverse_peaks.xls
I got a warning message saying that "Genometools binary" can't be called but I couldn't find the log file gt_gff3.log in the directory where I executed the run. From the warning message, it's a bit obscure whether anything wrong has happened or not:
2023-01-31 15:58:13,954 - INFO - Finished calling reverse strand peaks.
2023-01-31 16:03:12,669 - INFO - Finished calling forward strand peaks.
INFO Iterating over peaks to annotate 3' UTRs.: 535600it [04:29, 1986.50it/s]
2023-01-31 16:07:44,183 - INFO - Merging annotations with canonical gff file.
2023-01-31 16:07:45,163 - WARNING - Genometools binary can't be called. Please ensure it is installed.
2023-01-31 16:07:45,164 - WARNING - Some issues were encountered when processing output file. Check gt_gff3.log.
2023-01-31 16:07:45,172 - INFO - Writing summary statistics file.
2023-01-31 16:07:46,109 - INFO - peaks2utr finished successfully.
2023-01-31 16:07:47,112 - INFO - Clearing cache.
(For brevity, I only showed the relevant parts here. No warning messages elsewhere during the run.)
Let me show more details about the run:
gtf format (rather than gff) and I thought the warning message could have arisen because of the format incompatibility. So I went to convert my gtf file to gff3 and ran peaks2utr again, but the warning message persisted.peaks2utr and other necessary packages such as pandas installed) on a linux server.Thank you so much!
Hi,
With peaks2utr version 1.0.0, I got the error message:
2023-05-05 15:28:35,330 - DEBUG - CriteriaFailure - Peak forward_peak_10513 wholly contained within transcript ENSMUST00000149822
2023-05-05 15:28:35,331 - DEBUG - Peak forward_peak_10513 overlapping following gene ENSMUSG00000104740: Truncating
2023-05-05 15:28:35,334 - ERROR - Peak forward_peak_10513 produced abnormal 3' UTR <UTR: (117077435, 117077348)> for gene ENSMUSG00000088529. This is a bug, please report at https://github.com/haessar/peaks2utr/issues.
2023-05-05 15:28:35,338 - DEBUG - PEAK forward_peak_10513 CORRESPONDS TO 3' UTR <UTR: (117077457, 117077490)> OF GENE ENSMUSG00000104740
Here is plot of the region:

Installation of peaks2utr within a conda virtual environment with Python 3.10 seems to fail:
# Create a new virtual environment using conda
conda create --name peaks2utr
# Activate the newly created virtual environment
conda activate peaks2utr
conda install pip
This should have installed the default Python version. I went to check which python version by
python3 --version
which turns out to be
Python 3.10.8
Version of pip(pip3) is
pip 22.3.1 from ~/opt/miniconda3/envs/peaks2utr/lib/python3.10/site-packages/pip (python 3.10)
(where I manually replaced my home directory with ~)
Then I installed peaks2utr by typing
pip install peaks2utr
I got the following error messages:
Collecting peaks2utr
Using cached peaks2utr-0.2-py3-none-any.whl (14 kB)
Collecting pysam
Downloading pysam-0.20.0-cp310-cp310-macosx_10_9_x86_64.whl (3.2 MB)
━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 3.2/3.2 MB 9.4 MB/s eta 0:00:00
Collecting MACS2
Using cached MACS2-2.2.7.1.tar.gz (2.1 MB)
Installing build dependencies ... done
Getting requirements to build wheel ... error
error: subprocess-exited-with-error
× Getting requirements to build wheel did not run successfully.
│ exit code: 1
╰─> [1 lines of output]
CRITICAL: Python version must >= 3.6!
[end of output]
note: This error originates from a subprocess, and is likely not a problem with pip.
error: subprocess-exited-with-error
× Getting requirements to build wheel did not run successfully.
│ exit code: 1
╰─> See above for output.
note: This error originates from a subprocess, and is likely not a problem with pip.
The error message looks weird because I was indeed using Python>=3.6.
I also tried to install from source, but the same error message (from MACS2) persisted.
(Just a minor issue but the package build also needs to be installed in the newly created virtual environment for the "install from source" method).
Somehow, then I tried to create another virtual environment specifying the Python version to be used, and it finally worked this time (i.e., the installation was successful).
I specified Python 3.8 this time, as suggested by the manuscript uploaded to bioRxiv.
conda create --name peaks2utr_v2 python=3.8
conda activate peaks2utr_v2
Python version is:
Python 3.8.15
Hi Haessar,
Thanks for the awesome tool.
Here is a quick question with regards to an unexpected behavior of peaks2utr run where everything was on default setting except the --max-distance which I have set to 1500. I've shared two IGV screenshot below, where you can see the bam file I've used as input, and two gtf tracks, the top one is used as input, and the bottom one is the peaks2utr output where three_prime_utr has been added.
For the LOC111094256, although the alignment depth was not very deep, just 10 or so the tool added a three prime UTR.
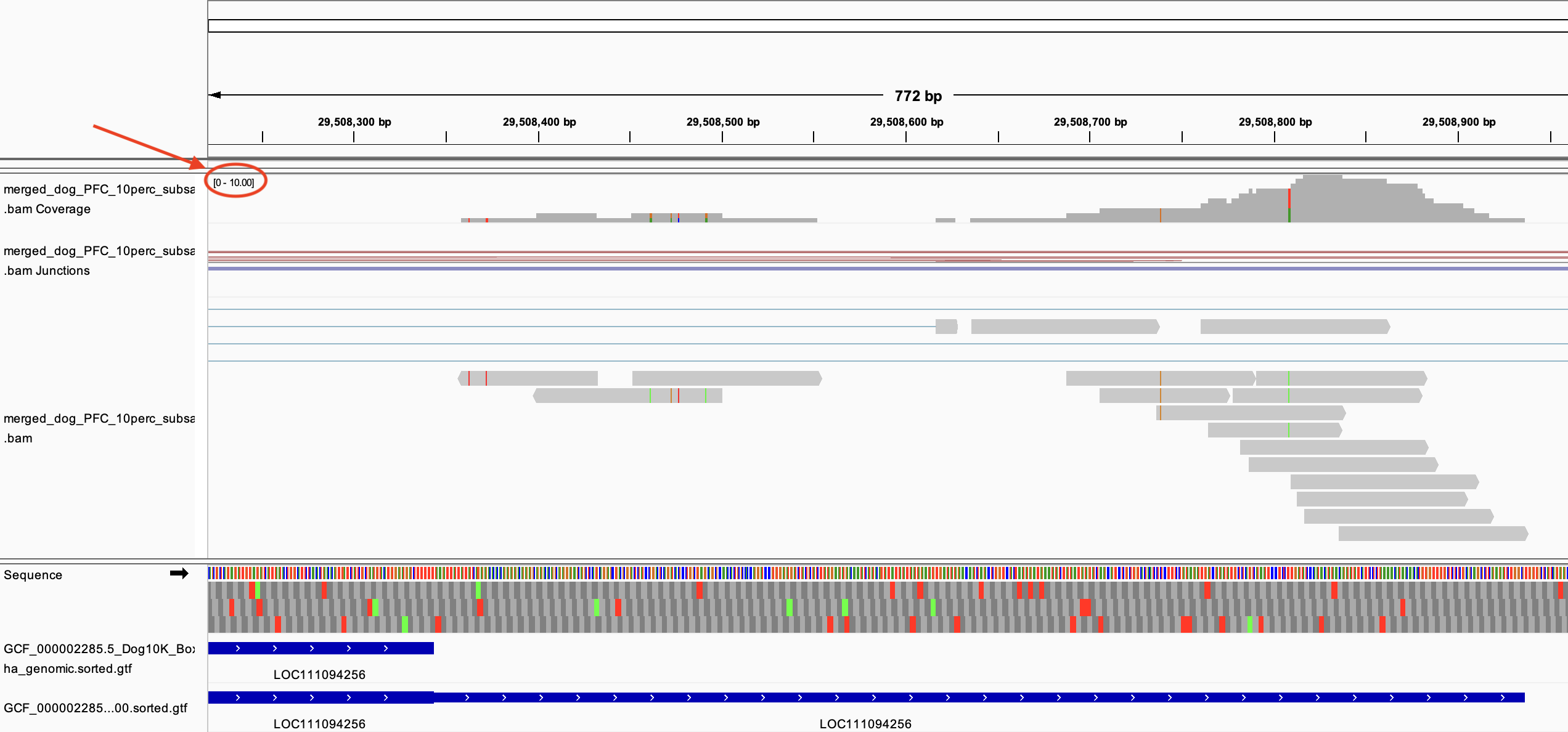
However, for GAD1, even though the alignment depth was much deeper (~200), the tool did not add a three prime UTR like I would expect.
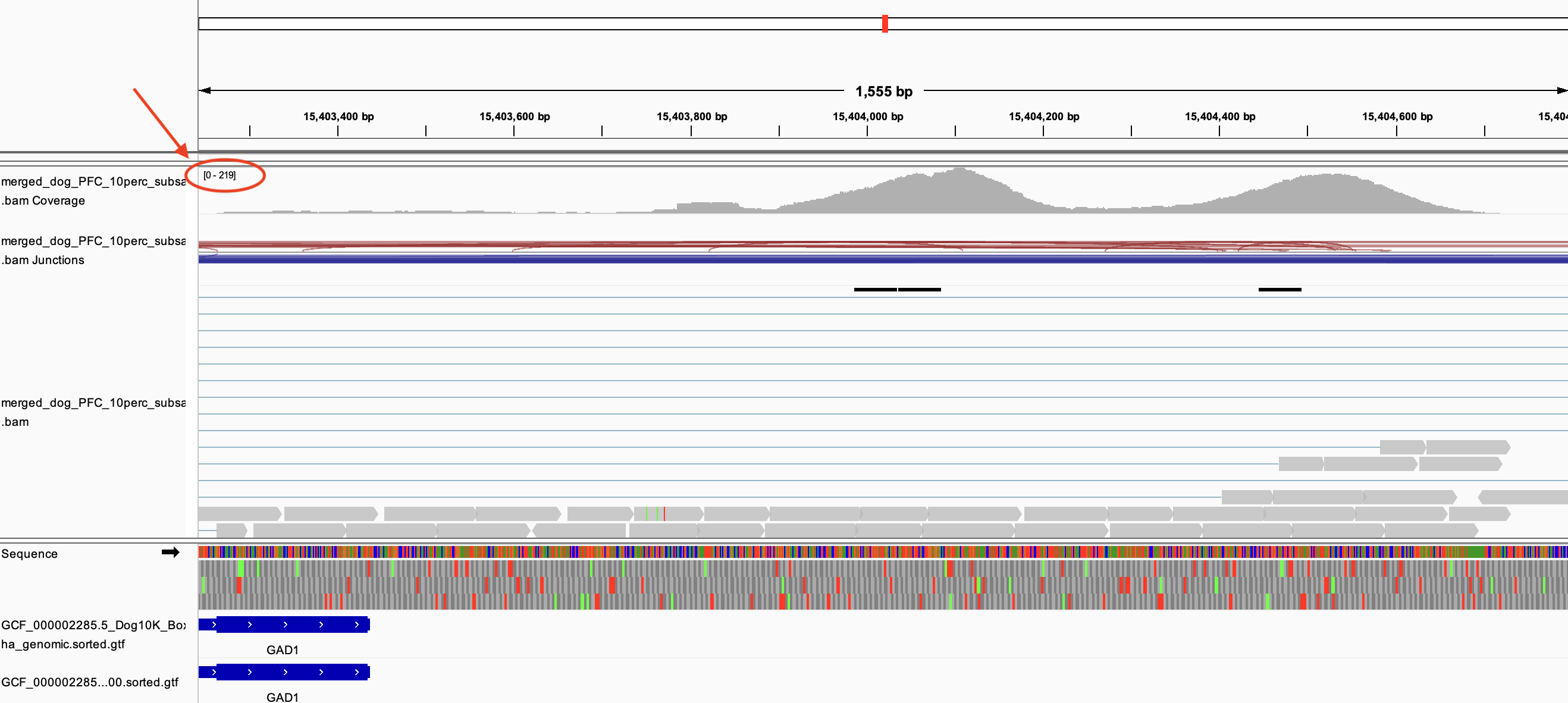
Why is this? What parameter could I tweak so that I can get these obvious three prime UTRs in my new gene annotations? Thank you! I am using 0.5.1.
Best,
JC
I wonder what the default for the option '--max-distance' is. We could not find any information about this in the command's help, publication, and documentation on GitHub. I would really appreciate your response.
Hi, I was testing whether the updated genome annotation file would lead to improved mapping statistics, so I tried to re-map my single-cell data with the updated genome annotation file. As CellRanger only accepts genome annotation files in GTF format (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/tutorial_mr), may I know which tool you would recommend us to use in order to convert the output from peaks2utr (which is in gff format) back to gtf?
I tried to use gffread but it seems the newly annotated 3'UTR would be either merged with exons or discarded (I guess something similar to the issue reported in gpertea/gffread#74 but I'm sorry that I am not so sure), which seems to add another layer of uncertainty to my comparison. So I guess it would be better to know what you think about this. Or perhaps there are alternative ways (like AGAT or other tools)? Looking forward to any advice - thanks so much.
Dear developer,
when I run peaks2utr, there is an error I can not solve, here is my log file:
2023-06-25 12:36:33,786 - INFO - Make .log directory
2023-06-25 12:36:33,787 - INFO - Make .cache directory
2023-06-25 12:36:33,787 - INFO - Splitting forward strand from out.bam.
2023-06-25 12:37:21,904 - INFO - Finished splitting forward strand.
2023-06-25 12:36:33,786 - INFO - Make .log directory
2023-06-25 12:36:33,787 - INFO - Make .cache directory
2023-06-25 12:36:33,787 - INFO - Splitting forward strand from out.bam.
2023-06-25 12:37:21,904 - INFO - Finished splitting forward strand.
2023-06-25 12:37:21,905 - INFO - Splitting reverse strand from out.bam.
2023-06-25 12:38:09,823 - INFO - Finished splitting reverse strand.
2023-06-25 12:38:09,828 - INFO - Merging SPAT outputs.
2023-06-25 12:38:09,828 - INFO - Filtering intervals with zero coverage.
[E::idx_find_and_load] Could not retrieve index file for '/home/jianyang/workspace/genome/L.dispar/annotation/07_utr/.cache/out.forward.bam'
[E::idx_find_and_load] Could not retrieve index file for '/home/jianyang/workspace/genome/L.dispar/annotation/07_utr/.cache/out.reverse.bam'
2023-06-25 12:47:39,324 - INFO - Creating gff db.
2023-06-25 12:47:39,325 - INFO - Calling peaks for forward strand with MACS3.
2023-06-25 12:47:39,333 - INFO - Calling peaks for reverse strand with MACS3.
2023-06-25 12:36:33,786 - INFO - Make .log directory
2023-06-25 12:36:33,787 - INFO - Make .cache directory
2023-06-25 12:36:33,787 - INFO - Splitting forward strand from out.bam.
2023-06-25 12:37:21,904 - INFO - Finished splitting forward strand.
2023-06-25 12:37:21,905 - INFO - Splitting reverse strand from out.bam.
2023-06-25 12:38:09,823 - INFO - Finished splitting reverse strand.
2023-06-25 12:38:09,828 - INFO - Merging SPAT outputs.
2023-06-25 12:38:09,828 - INFO - Filtering intervals with zero coverage.
[E::idx_find_and_load] Could not retrieve index file for '/home/jianyang/workspace/genome/L.dispar/annotation/07_utr/.cache/out.forward.bam'
[E::idx_find_and_load] Could not retrieve index file for '/home/jianyang/workspace/genome/L.dispar/annotation/07_utr/.cache/out.reverse.bam'
2023-06-25 12:47:39,324 - INFO - Creating gff db.
2023-06-25 12:47:39,325 - INFO - Calling peaks for forward strand with MACS3.
2023-06-25 12:47:39,333 - INFO - Calling peaks for reverse strand with MACS3.
2023-06-25 12:47:39,333 - INFO - Populating features
2023-06-25 12:47:39,333 - INFO - Populating features
Populating features table and first-order relations: 0 features^M2023-06-25 12:47:39,339 - INFO - Clearing cache.
Traceback (most recent call last):
File "/home/jianyang/mambaforge-pypy3/envs/utr/lib/python3.8/site-packages/gffutils/create.py", line 589, in _populate_from_lines
self._insert(f, c)
File "/home/jianyang/mambaforge-pypy3/envs/utr/lib/python3.8/site-packages/gffutils/create.py", line 530, in _insert
cursor.execute(constants._INSERT, feature.astuple())
sqlite3.IntegrityError: UNIQUE constraint failed: features.id
During handling of the above exception, another exception occurred:
Traceback (most recent call last):
File "/home/jianyang/mambaforge-pypy3/envs/utr/bin/peaks2utr", line 8, in
sys.exit(main())
File "/home/jianyang/mambaforge-pypy3/envs/utr/lib/python3.8/site-packages/peaks2utr/init.py", line 78, in main
asyncio.run(_main(args))
File "/home/jianyang/mambaforge-pypy3/envs/utr/lib/python3.8/asyncio/runners.py", line 43, in run
return loop.run_until_complete(main)
File "/home/jianyang/mambaforge-pypy3/envs/utr/lib/python3.8/asyncio/base_events.py", line 608, in run_until_complete
return future.result()
File "/home/jianyang/mambaforge-pypy3/envs/utr/lib/python3.8/site-packages/peaks2utr/init.py", line 158, in _main
db, _, _ = await asyncio.gather(
File "/home/jianyang/mambaforge-pypy3/envs/utr/lib/python3.8/site-packages/peaks2utr/preprocess.py", line 158, in create_db
await sync_to_async(gffutils.create_db)(
File "/home/jianyang/mambaforge-pypy3/envs/utr/lib/python3.8/site-packages/asgiref/sync.py", line 479, in call
ret: _R = await loop.run_in_executor(
File "/home/jianyang/mambaforge-pypy3/envs/utr/lib/python3.8/concurrent/futures/thread.py", line 57, in run
result = self.fn(*self.args, **self.kwargs)
File "/home/jianyang/mambaforge-pypy3/envs/utr/lib/python3.8/site-packages/asgiref/sync.py", line 538, in thread_handler
return func(*args, **kwargs)
File "/home/jianyang/mambaforge-pypy3/envs/utr/lib/python3.8/site-packages/gffutils/create.py", line 1292, in create_db
c.create()
File "/home/jianyang/mambaforge-pypy3/envs/utr/lib/python3.8/site-packages/gffutils/create.py", line 507, in create
self._populate_from_lines(self.iterator)
File "/home/jianyang/mambaforge-pypy3/envs/utr/lib/python3.8/site-packages/gffutils/create.py", line 591, in _populate_from_lines
fixed, final_strategy = self._do_merge(f, self.merge_strategy)
File "/home/jianyang/mambaforge-pypy3/envs/utr/lib/python3.8/site-packages/gffutils/create.py", line 226, in _do_merge
raise ValueError("Duplicate ID {0.id}".format(f))
ValueError: Duplicate ID cds.evm.model.chr2.1
How can I fix it?
Thanks
yours adoptbai
Problem when running on peaks2utr on gtf file
File "/home/jhc103/miniconda3/envs/jupyter-general/lib/python3.9/site-packages/gffutils/create.py", line 1292, in create_db
c.create()
File "/home/jhc103/miniconda3/envs/jupyter-general/lib/python3.9/site-packages/gffutils/create.py", line 507, in create
self._populate_from_lines(self.iterator)
File "/home/jhc103/miniconda3/envs/jupyter-general/lib/python3.9/site-packages/gffutils/create.py", line 591, in _populate_from_lines
fixed, final_strategy = self._do_merge(f, self.merge_strategy)
File "/home/jhc103/miniconda3/envs/jupyter-general/lib/python3.9/site-packages/gffutils/create.py", line 226, in _do_merge
raise ValueError("Duplicate ID {0.id}".format(f))
ValueError: Duplicate ID cds-XP_003638833.2
(toolshed-peaks2utr) [jhc103@tscc-9-14 sample2C-annotation]$ tail output_gtf_run_genometools.txt
result = self.fn(*self.args, **self.kwargs)
File "/home/jhc103/miniconda3/envs/jupyter-general/lib/python3.9/site-packages/asgiref/sync.py", line 490, in thread_handler
return func(*args, **kwargs)
File "/home/jhc103/miniconda3/envs/jupyter-general/lib/python3.9/site-packages/gffutils/create.py", line 1292, in create_db
c.create()
File "/home/jhc103/miniconda3/envs/jupyter-general/lib/python3.9/site-packages/gffutils/create.py", line 507, in create
self._populate_from_lines(self.iterator)
File "/home/jhc103/miniconda3/envs/jupyter-general/lib/python3.9/site-packages/gffutils/create.py", line 788, in _populate_from_lines
parent = f.attributes[self.transcript_key][0]
IndexError: list index out of range
Hi, sorry for having posted so many issues recently.
I was simply wondering whether it's possible to update the old genome annotation file with BAM files from multiple samples. I have multiple samples from the same species (which is a non-model organism), so what I tried was to update the genome annotation file with the BAM file from one single-cell sample at a time. However, when I tried to update the already-updated gff file with the BAM file from a different single-cell sample, I got the following error message (i.e., original gtf/gff → updated version 1 → updated version 2; it failed at the conversion step from updated version 1 to updated version 2) :
INFO Iterating over reads to determine SPAT pileups: 0%| | [20:53<?]
2023-02-01 22:28:29,771 - INFO - Merging SPAT outputs.
2023-02-01 22:28:30,058 - INFO - Creating gff db.
2023-02-01 22:28:30,060 - INFO - Calling peaks for forward strand with MACS2.
2023-02-01 22:28:30,069 - INFO - Calling peaks for reverse strand with MACS2.
2023-02-01 22:28:30,069 - INFO - Populating features
2023-02-01 22:28:30,069 - INFO - Populating features
2023-02-01 22:28:30,227 - INFO - Clearing cache.ons: 2000 features
Traceback (most recent call last):
File "/~~~home_dir~~~/tools/miniconda3/envs/peaks2utr/bin/peaks2utr", line 8, in <module>
sys.exit(main())
File "/~~~home_dir~~~/tools/miniconda3/envs/peaks2utr/lib/python3.9/site-packages/peaks2utr/__init__.py", line 49, in main
asyncio.run(_main())
File "/~~~home_dir~~~/tools/miniconda3/envs/peaks2utr/lib/python3.9/asyncio/runners.py", line 44, in run
return loop.run_until_complete(main)
File "/~~~home_dir~~~/tools/miniconda3/envs/peaks2utr/lib/python3.9/asyncio/base_events.py", line 647, in run_until_complete
return future.result()
File "/~~~home_dir~~~/tools/miniconda3/envs/peaks2utr/lib/python3.9/site-packages/peaks2utr/__init__.py", line 102, in _main
db, _, _ = await asyncio.gather(
File "/~~~home_dir~~~/tools/miniconda3/envs/peaks2utr/lib/python3.9/site-packages/peaks2utr/preprocess.py", line 120, in create_db
await sync_to_async(gffutils.create_db)(gff_in, gff_db, force=True, verbose=True, disable_infer_genes=True, disable_infer_transcripts=True)
File "/~~~home_dir~~~/tools/miniconda3/envs/peaks2utr/lib/python3.9/site-packages/asgiref/sync.py", line 448, in __call__
ret = await asyncio.wait_for(future, timeout=None)
File "/~~~home_dir~~~/tools/miniconda3/envs/peaks2utr/lib/python3.9/asyncio/tasks.py", line 442, in wait_for
return await fut
File "/~~~home_dir~~~/tools/miniconda3/envs/peaks2utr/lib/python3.9/concurrent/futures/thread.py", line 58, in run
result = self.fn(*self.args, **self.kwargs)
File "/~~~home_dir~~~/tools/miniconda3/envs/peaks2utr/lib/python3.9/site-packages/asgiref/sync.py", line 490, in thread_handler
return func(*args, **kwargs)
File "/~~~home_dir~~~/tools/miniconda3/envs/peaks2utr/lib/python3.9/site-packages/gffutils/create.py", line 1292, in create_db
c.create()
File "/~~~home_dir~~~/tools/miniconda3/envs/peaks2utr/lib/python3.9/site-packages/gffutils/create.py", line 507, in create
self._populate_from_lines(self.iterator)
File "/~~~home_dir~~~/tools/miniconda3/envs/peaks2utr/lib/python3.9/site-packages/gffutils/create.py", line 589, in _populate_from_lines
self._insert(f, c)
File "/~~~home_dir~~~/tools/miniconda3/envs/peaks2utr/lib/python3.9/site-packages/gffutils/create.py", line 530, in _insert
cursor.execute(constants._INSERT, feature.astuple())
sqlite3.InterfaceError: Error binding parameter 11 - probably unsupported type.
The code I ran was:
peaks2utr --extend-utr -p 25 -o <GENOME_ANNOTATION>.update2.gff3 /path/to/<GENOME_ANNOTATION>.update1.gff3 /path/to/the/second/replicate/sample/outs/possorted_genome_bam.bam
Thank you so much
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.