bzhanglab / pepquery Goto Github PK
View Code? Open in Web Editor NEWPepQuery: a targeted peptide search engine
Home Page: http://pepquery.org
License: GNU General Public License v3.0
PepQuery: a targeted peptide search engine
Home Page: http://pepquery.org
License: GNU General Public License v3.0










What is the meaning of -1 in some of the rows in the n_ptm, n_random, and total_random of the output file psm_rank.txt?
Thank you
Will you consider to add unspecific digestion for immunopeptidomics identifications?
Many thanks,
Eden.
The documentation clearly describes some of the files produced by pepquery following the search of a peptide in an mgf dataset. However, it also produces a few files for which I could not find a description in the documentation. For example, the files psm.txt and detail.txt.
Could you also indicate what the absence of the ptm_detail.txt file means? I often get a ptm.txt file without a detail_ptm.txt file.
I run pepquery using peptide search with unrestricted modification and our custom database of reference proteins.
Thank you
We have been running pepquery 1 and 2 with exactly the same parameters and noticed difference between the obtained results. Some spectra appeared confident with version 1 and are no longer confident in version 2. We noticed that this is due to the n_ptm value that is getting larger numbers in version 2. We understand from reading issue #45 this could be due to the updates in version 2 considering multiple variable modifications in the unrestricted modifications searching step vs only 1 in version 1. This means that more peptides are considered and this might increase this n_ptm number.
Do you consider a very stringent cutoff is needed when working with novel sequences or do you think we can set n_ptm to be smaller than a certain value (such as 3 or 5 maybe) to have a less stringent cutoff? In that case do you have any recommendations on the threshold to use?
Hi Bo,
I was wondering if there is a way to include TMT reporter tags (e.g. 126, 127n, ... 130c, 131) in addition to the "TMT 10-plex of peptide N-term"? I have run some analysis on the CCLE proteome dataset but can only narrow down the peptide to a particular 10-plex experiment, not the specific cell line/sample.
(Apologies if this is off topic, this field is new to me)
Thank you,
Andrea
Hello,
To create the ms database for the CCLE_proteome_MSV000085836 data did you use the commands as described in the instructions page:
msconvert --filter "peakPicking true 1-2" --mgf *.raw
then java -cp pepquery-1.4.jar main.java.index.BuildMSlibrary
or were there additional steps or arguments?
Thank you,
Andrea
I am running pepquery 1.2.0 on an HPC platform using many cpus to search many mgf files at the same time. Sometimes the process completes with no problems but many of them show the following message and get stuck:
...
Finished proteins:140000
Finished proteins:141000
Protein sequences:141748
2019-10-25 14:27:24 [ INFO] Time elapsed: 2.23 min
Peptides with matched spectra: 2
pool-3-thread-1 -> Start 1: TDTMEYFISPRCDMDYFSLGHKCLQQLVFDR ...
pool-3-thread-2 -> Start 2: GQSLRPCTWLPEEWVLELLCTDTDLEERQGR ...
pool-3-thread-2 -> Finished 2: GQSLRPCTWLPEEWVLELLCTDTDLEERQGR .
pool-3-thread-1 -> Finished 1: TDTMEYFISPRCDMDYFSLGHKCLQQLVFDR .
All spectra have been exported to file: /nfs3_ib/ip32-ib/home/sleblanc/PepQ_BCAC_snpeff/TCGA_A2-A0YF_BH-A0DD_BH-A0E9_117C_P_BI_20131104_H-PM_f04//psm_rank.mgf
The number of spectra that need to be validated by PTM searching: 1
SLF4J: Class path contains multiple SLF4J bindings.
SLF4J: Found binding in [jar:file:/home/group/tools/PepQuery_v1.2.0/lib/slf4j-log4j12-1.6.1.jar!/org/slf4j/impl/StaticLoggerBinder.class]
SLF4J: Found binding in [jar:file:/home/group/tools/PepQuery_v1.2.0/lib/logback-classic-1.0.11.jar!/org/slf4j/impl/StaticLoggerBinder.class]
SLF4J: See http://www.slf4j.org/codes.html#multiple_bindings for an explanation.
SLF4J: Actual binding is of type [org.slf4j.impl.Log4jLoggerFactory]
All modifications in unimod:1375
Use enzyme:Trypsin
What should I do about this?
Hi Bo,
Sorry for the question which is an off-topic and directly not related to pepquery. I doing some peptide identifications in colon prospective PNNL dataset and would like to match my PSM to the actual patient ID - in order to correlate proteome to the genomics. I found it's hard to trace down the sample labels in PNNL dataset to the actual patient. For example, my search gave the following output:
SRQMESLGMK | TMT 10-plex of peptide N-term@0[229.1629];TMT 10-plex of K@10[229.1629] | 30 | 3 | 1,623.888 | -2.562 | 1,623.884 | 542.303 | 21.706 | 0 | 118 | 1 | 995 | 0.002008 | 1 | 0 | Retained | Yes | 09CPTAC_COprospective_W_PNNL_20170123_B3S1_f08:15737:3
however, "09CPTAC_COprospective_W_PNNL_20170123_B3S1_f08" does not match any patient id in supplemental data for Cell 2019 CPTAC paper or metadata stored on CPTAC server.
Thank you for your help,
##Vladimir
Hi, I was working on some WES(WXS) data and mzml data. Basicly i follewed the Variant Peptide Identification procedure in this paper 《Integrated Proteogenomic Characterization of Clear
Cell Renal Cell Carcinoma》, use custmdbj to creat Patient-Specific Protein Sequence Database, use MSGF+ to search and use Pepquery to validation novel peptides and use PDV to visualization.
Could i know how you annotating these peptides in psm_rank.txt.
Because i tried to map psms in psm_rank.txt back to annotated vcf file, but one peptide squence can match more than one protein sequence in custombdj created Patient-Specific Protein Sequence Database, and one protein sequence matched in hg19_refGene.txt could match more than one mutated position in vcf file. I did not figure out how to wipe off these false positive.
I am using PepQuery to validate novel peptides and I came across the following unexpected behavior:
My reference db contains the following two sequences:
sp|P50395|GDIB_HUMAN Rab GDP dissociation inhibitor beta OS=Homo sapiens OX=9606 GN=GDI2 PE=1 SV=2
...TY_MLNKPIEEIIVQNGK_V...
and
sp|P06748|NPM_HUMAN Nucleophosmin OS=Homo sapiens OX=9606 GN=NPM1 PE=1 SV=2
...FK_VDNDENEHQLSLR_TVS...
and my query file contains the following peptides:
MLNKPIEEIIVQDGK
VDNDEDEHQLSLR
I run the PepQuery search using the following command:
java -jar pepquery-1.4.1/pepquery-1.4.1.jar \
-itol 0.05 -hc TRUE -um -cpu 4 -pep search_files/q10767_search.txt \
-db swissprot_UP000005640.fasta -ms spectra/q10767.mgf -o pepq_test/
As expected, PSMs with VDNDEDEHQLSLR are not confident because it is one deamidation away from a peptide in the above NPM1 reference and thus receives an equal score.
However, for MLNKPIEEIIVQDGK , we get a confident association to a spectrum even though it is a peptide one deamidation away from a GDI2 peptide as shown above. We expected the same output as with NPM1 and can seem to find an explanation. Could it be because our peptide contains a miss-cleavage? Is there some other explanation?
Thank you!
Hello, the first step of a search always involves indexing the reference library into a sql db. Is it possible to make this persist for future searches?
thank you
Hello,
I would like to use the PepQuery website, but whenever I click on the link, I get an error message ("aws2.zhang-lab.org took too long to respond.") This problem occurs on all computers and browsers we have tried.
Do you have any idea what the problem might be?
Thank you for your help!
Is it possible to add another PTM which is not in the current list of 159 PTM ?
I would need the unimod 213: ADP Ribose addition
C(15)H(21)N(5)O(13)P(2)
mass: 541.06110901175
site: S
That PTM is mostly found on Serine, but can also be found on other amino acid (PMID : 30157440)
Thanks
Hello,
I am new to the proteomics and would like to use pepquery to different MS datasets. In the list of standalone pepquery parameters, "-itol" seems to be machine-dependent. Which value should I provide for each MS machine currently available on the market?
Thank you
I have some peptides I identify from an MS/MS analysis. I would like to know just the hyperscore and MVH related to its sequence spectrum (which I already have)...is there way to get jut those scores through your stand-alone version? Thank you.
Hello,
Is it valuable to apply hyper score threshold to filter the most significant PSM spectra?
Let's say, I get ~3000 PSMs with p-value < 0.01. Will it be useful to stringen selection with hyper score threshold? If yes, which one to apply?
Hi @wenbostar,
I try to run pepquery2 on the phosphoproteome data, but it didn’t finish running after several hours, even for only one peptide. Could you please help to check about the following running log? Thanks.
java -jar -Xmx40G ~/Programs/pepquery-2.0.2/pepquery-2.0.2.jar -e 1 -c 2 -db DB/refprot.fa -fixMod 1,11,12 -varMod 2,5,7,8,9 -maxVar 3 -cpu 20 -fragmentMethod 1 -tolu ppm -tol 20 -itol 0.05 -minLength 7 -maxLength 45 -ms S063_LSCC_Satpathy_Cell2021_Phosphoproteome/01CPTAC_LSCC_Phosphoproteome_BI_20190619/pepquery2.BuildMSlibrary/ -m 1 -minScore 12 -n 10000 -hc -o testPQ2/ -i AGGTTAMHRGLIHPQSRAVGGDAMTPILTVLICLGLSLGPR
2023-04-11 16:30:00 [INFO ] main.java.pg.CParameter[updateCParameter:873] - Task type: novel peptide identification
2023-04-11 16:30:00 [INFO ] uk.ac.ebi.pride.utilities.pridemod.io.unimod.xml.unmarshaller.UnimodUnmarshallerFactory[initializeUnmarshaller:41] - Unmarshaller Initialized
2023-04-11 16:30:00 [INFO ] main.java.OpenModificationSearch.ModificationDB[importPTMsFromUnimod:355] - All modifications in unimod:1375
2023-04-11 16:30:00 [INFO ] main.java.pg.PeptideSearchMT[search:243] - Start analysis
#############################################
PepQuery parameter:
2023-04-11 16:30:00 [INFO ] main.java.pg.DatabaseInput[getEnzymeByIndex:263] - Use enzyme:Trypsin
PepQuery version: 2.0.2
PepQuery command line: -e 1 -c 2 -db DB/refprot.fa -fixMod 1,11,12 -varMod 2,5,7,8,9 -maxVar 3 -cpu 20 -fragmentMethod 1 -tolu ppm -tol 20 -itol 0.05 -minLength 7 -maxLength 45 -ms S063_LSCC_Satpathy_Cell2021_Phosphoproteome/01CPTAC_LSCC_Phosphoproteome_BI_20190619/pepquery2.BuildMSlibrary/ -m 1 -minScore 12 -n 10000 -hc -o testPQ2/ -i AGGTTAMHRGLIHPQSRAVGGDAMTPILTVLICLGLSLGPR
Fixed modification: 1,11,12 = Carbamidomethylation of C,TMT 10-plex of K,TMT 10-plex of peptide N-term
Variable modification: 2,5,7,8,9 = Oxidation of M,Acetylation of peptide N-term,Phosphorylation of S,Phosphorylation of T,Phosphorylation of Y
Max allowed variable modification: 3
Add AA substitution: false
Enzyme: 1 = Trypsin
Max Missed cleavages: 2
Precursor mass tolerance: 20.0
Range of allowed isotope peak errors: 0
Precursor ion mass tolerance unit: ppm
Fragment ion mass tolerance: 0.05
Fragment ion mass tolerance unit: Da
Scoring algorithm: 1 = Hyperscore
Min score: 12.0
Min peaks: 10
Min peptide length: 7
Max peptide length: 45
Min peptide mass: 500.0
Max peptide mass: 10000.0
Random peptide number: 10000
Fast mode: false
CPU: 20
#############################################
2023-04-11 16:30:01 [INFO ] main.java.pg.PeptideSearchMT[search:250] - Spectrum ID type:1, use 1-based number as index for a spectrum.
2023-04-11 16:30:01 [INFO ] main.java.pg.PeptideSearchMT[search:253] - Step 1: target peptide sequence preparation and initial filtering ...
2023-04-11 16:30:01 [INFO ] main.java.pg.PeptideSearchMT[search:330] - Input peptide sequences:1
2023-04-11 16:30:01 [INFO ] main.java.pg.InputProcessor[searchRefDB:413] - Don't find indexed database:DB/refprot.fa.sqldb
2023-04-11 16:30:01 [INFO ] main.java.pg.InputProcessor[searchRefDB:414] - Use database:DB/refprot.fa
2023-04-11 16:30:01 [INFO ] main.java.pg.PepMapping[init:62] - Load db file: DB/refprot.fa
2023-04-11 16:30:01 [INFO ] main.java.pg.PepMapping[init:78] - Start indexing fasta file
2023-04-11 16:30:05 [INFO ] main.java.pg.PepMapping[init:88] - Indexing took 0.994814493 seconds and consumes 175.904832 MB
2023-04-11 16:30:05 [INFO ] main.java.pg.PeptideSearchMT[search:407] - Valid target peptides: 1
2023-04-11 16:30:05 [INFO ] main.java.pg.PeptideSearchMT[generatePeptideInputs:1330] - Generate peptide objects ...
2023-04-11 16:30:05 [INFO ] main.java.pg.PeptideSearchMT[generatePeptideInputs:1341] - CPU: 1
2023-04-11 16:30:05 [INFO ] main.java.pg.PeptideSearchMT[generatePeptideInputs:1359] - Generate peptide objects done.
2023-04-11 16:30:05 [INFO ] main.java.pg.PeptideSearchMT[search:413] - Step 1: target peptide sequence preparation and initial filtering done: time elapsed = 0.09 min
2023-04-11 16:30:05 [INFO ] main.java.pg.PeptideSearchMT[search:418] - Step 2: candidate spectra retrieval and PSM scoring ...
2023-04-11 16:30:05 [INFO ] main.java.pg.PeptideSearchMT[search:421] - Input for MS/MS data is a folder (MS/MS library):S063_LSCC_Satpathy_Cell2021_Phosphoproteome/01CPTAC_LSCC_Phosphoproteome_BI_20190619/pepquery2.BuildMSlibrary/
2023-04-11 16:30:05 [INFO ] main.java.pg.SpectraInput[readSpectraFromMSMSlibrary:371] - Used CPUs: 1
2023-04-11 16:30:05 [WARN ] main.java.msio.MsLibrarySearchWorker[loadSpectra:201] - File doesn't exist:S063_LSCC_Satpathy_Cell2021_Phosphoproteome/01CPTAC_LSCC_Phosphoproteome_BI_20190619/pepquery2.BuildMSlibrary//42683.mgf
2023-04-11 16:30:05 [WARN ] main.java.msio.MsLibrarySearchWorker[loadSpectra:201] - File doesn't exist:S063_LSCC_Satpathy_Cell2021_Phosphoproteome/01CPTAC_LSCC_Phosphoproteome_BI_20190619/pepquery2.BuildMSlibrary//45035.mgf
2023-04-11 16:30:05 [WARN ] main.java.msio.MsLibrarySearchWorker[loadSpectra:201] - File doesn't exist:S063_LSCC_Satpathy_Cell2021_Phosphoproteome/01CPTAC_LSCC_Phosphoproteome_BI_20190619/pepquery2.BuildMSlibrary//46634.mgf
2023-04-11 16:30:06 [WARN ] main.java.msio.MsLibrarySearchWorker[loadSpectra:201] - File doesn't exist:S063_LSCC_Satpathy_Cell2021_Phosphoproteome/01CPTAC_LSCC_Phosphoproteome_BI_20190619/pepquery2.BuildMSlibrary//43163.mgf
2023-04-11 16:30:06 [INFO ] main.java.pg.SpectraInput[readSpectraFromMSMSlibrary:393] - Matched spectra: 19
2023-04-11 16:30:06 [INFO ] main.java.pg.PeptideSearchMT[search:456] - Time elapsed: 0.11 min
2023-04-11 16:30:06 [INFO ] main.java.pg.PeptideSearchMT[search:476] - Step 2: candidate spectra retrieval and PSM scoring done: time elapsed = 0.11 min
2023-04-11 16:30:06 [INFO ] main.java.pg.PeptideSearchMT[search:480] - Step 3-4: competitive filtering based on reference sequences and statistical evaluation ...
2023-04-11 16:30:06 [INFO ] main.java.pg.PeptideSearchMT[search:515] - Don't find indexed database:DB/refprot.fa.sqldb
2023-04-11 16:30:06 [INFO ] main.java.pg.PeptideSearchMT[search:522] - Use database:DB/refprot.fa
2023-04-11 16:30:06 [INFO ] main.java.pg.DatabaseInput[getEnzymeByIndex:263] - Use enzyme:Trypsin
2023-04-11 16:30:43 [INFO ] main.java.pg.DatabaseInput[protein_digest:474] - Protein sequences:132437, total unique peptide sequences:2996576
2023-04-11 16:30:43 [INFO ] main.java.pg.DatabaseInput[protein_digest:475] - Time used for protein digestion:36 s.
Hello,
I am running web server version of PepQuery using the following input parameters:
MS/MS dataset: Prospective_Colon_VU
Target event: Protein Sequence
Input sequence: ".."
Reference database: "Refseq"
Scoring algorithm: MVH
Perform unrestricted modification filtering: yes.
and I receive an error:
"Error message: simpleWarning in system(javabin, intern = TRUE): running command 'java -Xmx30G -jar tool/proteinID-1.0-SNAPSHOT.jar -cpu 0 -db data/refdb/hg38_RefSeq_20190910_eTrypsin_c1_f6_v117/hg38_RefSeq_20190910.fasta -fixMod 6 -fragmentMethod 1 -itol 0.05 -tol 10 -tolu ppm -varMod 117 -ms s3://pdc-pepquery/cptac_colon_2019_labelfree/ -minScore 12 -maxVar 3 -c 1 -m 1 -i RSNSKKKGRRNRIPAVLRTEGEPLHTPSVGMRETTGLGC -t 1 -prefix Wed_Mar_25_210050_2020_protein -o result/Wed_Mar_25_210050_2020_protein/ -n 1000 -um' had status 1"
Is there anything I should apply to run the request successfully?
Thank you!
Vladimir
Hi developers of PepQuery,
thank you for this very nice tool. We tried it and with the help of the good instruction it works very nice! I have two question:
about the Signal-to-Noise-ratio: Is there any cut-off value (e.g. 0.1 % intensity of the base peak) for the search with PepQuery? Is it possible to manipulate this? The same question is for your visualisation tool PDV: Is there a possibility to manipulate the intensity threshold when a peak is annotated?
about the search with multiple raw data files: Because mgf-files are simple text files, is it possible to connect the different mgf-files manually and search against this combined file? Or is there anything why this is not a good idea? Unfortunately, the "msdatasets.json"-file in the documentation is not invocable.
Thank you very much in advance for your answer!
Best regards,
I ran the stand-alone version of Pepquery. As a test, I modified the iPRG one peptide search example for human as follows:
java -jar -Xmx2G ../pepquery.jar -db vcf/Ref_Hsapiens_GRCh37/hg19_refGenePro.fa -fixMod 6 -varMod 107,142,143,15 -maxVar 3 -cpu 4 -fragmentMethod 1 -itol 0.02 -tol 10 -ms vcf/TCGA-A6-3807-01A_f0106.mgf -pep AVVQDPALKPLALVYGEATSR -m 1 -minScore 10 -n 1000 -um -o out
How can I resolve the following error:
PSMs need to map to reference database:0
Exception in thread "main" java.lang.IndexOutOfBoundsException: Index -1 out of bounds for length 0
at java.base/jdk.internal.util.Preconditions.outOfBounds(Preconditions.java:64)
at java.base/jdk.internal.util.Preconditions.outOfBoundsCheckIndex(Preconditions.java:70)
at java.base/jdk.internal.util.Preconditions.checkIndex(Preconditions.java:248)
at java.base/java.util.Objects.checkIndex(Objects.java:372)
at java.base/java.util.ArrayList.get(ArrayList.java:458)
at main.java.pg.DatabaseInput.prepareSpectrumForPeptideSearch(DatabaseInput.java:541)
at main.java.pg.DatabaseInput.readDB(DatabaseInput.java:324)
at main.java.pg.PeptideSearchMT.main(PeptideSearchMT.java:461)
Hi, in the standalone version what does it mean for the the option for -e 0:Non enzyme, does it mean its pepquery its going to be looking for whole proteins or its an unspecified search?
I work with ancient proteins and I don't digest as they are heavily degraded any way, thus I do unspecific searches rather than specific ones or whole protein searches.
Hello,
I want to search on multiple MS/MS datasets, but json file for parameter -c of search_multiple_datasets.py (http://pepquery.org/data/msdatasets.json) is not available now . Besides, example file under multiple datasets (http://pepquery.org/data/msdatasets.tar.gz) is also not available. I get following error message for them: "You don't have permission to access this resource."
Is there any alternative link for these files. Can you help me?
Hi, Bo:
I tried both TCGA_Breast_cancer_proteome and TCGA_Colon_cancer_proteome. When I selected row in Identification Result, the Sample Information is empty.
As for TCGA_breast_cancer_proteome, results in modification column says iTRAQ 4-plex. There seems to be multiple sample names in spectrum title. How will you display sample information? How do I know the query peptide in which sample?
thanks for your time
ww

I am using the unrestricted modifications feature of pepquery and I would like to filter out some of the modifications based on their unimod classification. I am using the unimod xml to find the modification's specificity and classification but I am not sure what field to use to associate them with the pepquery output. I am mostly interested in the classes "post transnational modification" and "artifact", do you know how I can identify these in the pepquery output?
Thank you
Hello,
just stumbled over the difference between web-server and standalone version results. Seems, that web-server "target event" is not clearly defined in stand-alone version. What would be the parameters look like for st-alone pepquery, if I want to use whole protein sequence to search for peptide PSMs?
Thanks
Hi,
I have a similar question as Andrea- ie, to find the sample information based on iTRAQ labelling in TCGA BC dataset (instead of Andrea's CCLE analysis);
https://proteomic.datacommons.cancer.gov/pdc/study/PDC000111
The novel peptides that I find with PEPQUERY, I wish to map back to sample. I see it is possible using spectrum ID- but struggling to find spectrum information.
Sorry for being naive- I am very new to this field.
How can I find matching spectrum in this? Any help would be really appreciated.
Thank you very much in advance.
Abhijeet Pataskar
Hi, I am trying to detect a mutant protein that has W->F or W->M substitution. Now, I want to detect both of them using PEPQUERY and see which one is there in cancer proteomes (CPTAC). But, M-oxidation has the same mass as F. I wonder how does PEPQUERY handle this sameness in mass. When I detect W->F, I want to control that with W-> Moxi as well to be 100% sure it is W->F. Can PEPQUERY be used for this purpose?
Looking forward to hear from you.
Thank you :)
Abhijeet Pataskar
Hi, I have some general question about the application of PepQuery for the validation of novel peptides.
I was wondering if it is useful to use the unrestricted modification search for a organism, which had only about ~3000 predicted genes. Moreover, compared with human proteome, the proteome of a bacteria organism should be much less complex due to less modifications. I was afraid if it will increase false negative results, if we use unrestricted modifications. Do you think for bacteria it is useful to use PepQuery with open modification search for small bacteria proteoms to validate new peptides?
Just to clarify: What does “unrestricted modification search” means? Are this really all possible modifications (with all mass shifts) or are only the PTM list with the 159 PTMs considered? Do you think (in particular for organism with less expected posttranslational modifications, e.g. bacteria) it is useful to do a “restricted modification search” with for example the 20 most abundant known modifications + artefacts ?
It is for complex proteome samples very usual to have co-isolated peptides in the MS2 spectra. How PepQuery deals with theses spectra? For example, if a “new” peptide co-eluted with a peptide from a reference protein (with a high isolation interference of maybe 70 %) the spectra will potentially better match to the reference peptide and not to the new peptide. Will PepQuery reject the “new peptide”, because the reference peptide matched better to the spectra (despite this is due to co-elution)?
The Number of random peptides generated due to the p-value calculation is dependent on the peptide size. Is this a problem for short peptides (6-8 AS) (is there the p-Value calculation reliable?)?
Dear PepQuery2 developers,
Thanks for developing this powerful search engine. In your recent paper, PepQuery2 was used to query hundreds of nuORFs against proteomic datasets. We has also annotated thousands of novel ORFs recently and we would like to search them for MS evidence. I have reviewed the documentation page and I see that it is only possible to search a single novel protein at a time. I would be grateful if you could let me know if there is a way to search multiple novel proteins with PepQuery2.
Thank you for your time and consideration.
Sincerely,
Hong
Dear Team,
I tried to search for a peptide SRPMTVPSIDDYGR in the CCRCC_Phosphoproteome, but I got different results from pepquery online, and there are many such cases. How did this difference happen and how to improve the true positive rate of the results and get rid of the unreliable results?Thanks a lot.
My result:
charge 3
confident Yes
exp_mass 1981.82
modification TMT 10-plex of peptide N-term@0[229.1629];Phosphorylation of T@5[79.9663];Phosphorylation of S@8[79.9663]
mz 661.614
n 1
n_db 0
n_ptm 0
n_random 1
pep_mass 1981.86
peptide SRPMTVPSIDDYGR
ppm 18.1515
pvalue 0.001998
rank 1
score 12.1771
spectrum_title 10CPTAC_CCRCC_P_JHU_20180123_LUMOS_f02:8038:3
total_db 9
total_random 1000
Pepquery Online:
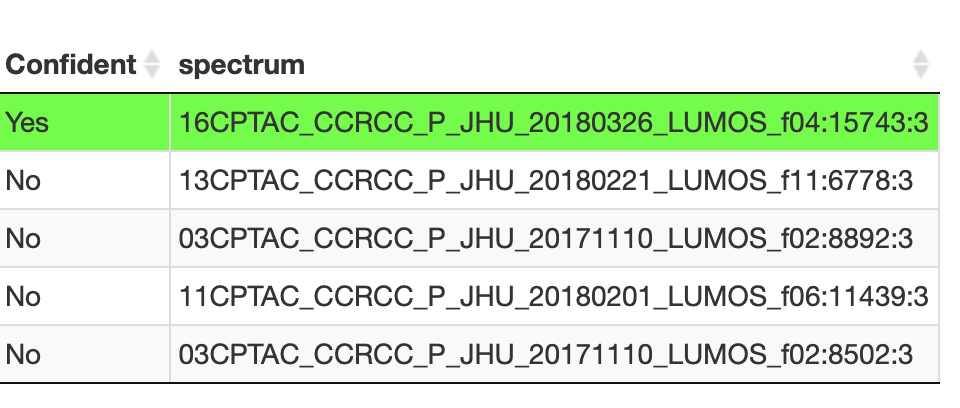
My param:
java -Xmx64G -jar pepquery-1.6.2/pepquery-1.6.2.jar -o cptac_out/${input}/${search} -cpu 16 -fixMod 6,62,108 -varMod 21,114,117,118,128 -tol 20 -tolu ppm -um -itol 0.05 -prefix pepqout -ms ${pepin} -pep ${fasta} -n 1000 -db 2020-11-26-reviewed-contam-UP000005640.fa -maxLength 45 -minLength 7 -maxVar 4 -indexType 1
It is wonderful that both online and standalone version of PepQuery can search TMT datasets. However, it occurred to me that PepQuery did not provide the info regarding which sample the results are matched to.
It seems that PepQuery did not provide TMT tag or reporter iron information in the output, and displayed the results based on TMT fractions. Is it possible to obtained the sample information for the peptide in the output? For example,I searched the peptide with GTEx_32_Tissues dataset, the peptide was found with spectrum_title named “Instrument1_sample01_121115_Fr01:17660:2”,but I could not exactly know it come from which GTEx Tissue.
@wenbostar and @bingzhang16
We have been testing the implementation of PepQuery2 in Galaxy and we have some results shared here.
In particular, PepQuery2 seems to generate less number of valid peptides as compared to the earlier version.
Tools |PSMs|Valid-PSMs|Confident Peptides
PepQuery1 |692|55|38
PepQuery2 |488|32|21
Please see a history that has the inputs (Datasets#1-3), and outputs from PepQuery1 (Datasets#4-7) and PepQuery2 (Datasets#8-16).
(https://usegalaxy.eu/u/pratikjagtap/h/pepquery2-test)
We were interested to know if the scoring method in PepQuery2 has been updated to make it more stringent and whether there have been any changes to the amino acid substitutions and PTM scoring.
PSM Rank Comparison.xls
I have meet some problem with indexed database.The command to run is as follows.Start indexing fasta file 50% has an error.
Looking forward to your reply,thanks
java -jar ./PepQuery_v1.3.0/pepquery-1.3.jar -pep EGGGGQPPARPATPSGR -db /hwfssz1/ST_CANCER/POL/SHARE/DataBase/Ensembl/Homo_sapiens.GRCh38.pep.all.fa -ms ./pwiz/TCGA_AO-A12D-01A_C8-A131-01A_AO-A12B-01A_Proteome_BI_20130208_MGF/TCGA_AO-A12D_C8-A131_AO-A12B_117C_W_BI_20130208_H-PM_f01.mgf
2019-10-19 16:53:45 [ INFO] Start analysis
2019-10-19 16:53:45 [DEBUG] -pep EGGGGQPPARPATPSGR -db /hwfssz1/ST_CANCER/POL/SHARE/DataBase/Ensembl/Homo_sapiens.GRCh38.pep.all.fa -ms ./pwiz/TCGA_AO-A12D-01A_C8-A131-01A_AO-A12B-01A_Proteome_BI_20130208_MGF/TCGA_AO-A12D_C8-A131_AO-A12B_117C_W_BI_20130208_H-PM_f01.mgf
#############################################
PepQuery parameter:
Fixed modification: 6 = Carbamidomethylation of C
Variable modification: 117 = Oxidation of M
Max allowed variable modification: 3
Add AA substitution: false
Enzyme: 1
Max Missed cleavages: 2
Precursor mass tolerance: 10.0
Precursor ion mass tolerance unit: ppm
Fragment ion mass tolerance: 0.6
Fragment ion mass tolerance unit: Da
Scoring algorithm: 1 = Hyperscore
Min score: 12.0
Min peaks: 10
Min peptide length: 7
Max peptide length: 45
Min peptide mass: 500.0
Max peptide mass: 10000.0
Random peptide number: 10000
CPU: 6
#############################################
Input peptide sequences:1
Don't find indexed database:/hwfssz1/ST_CANCER/POL/SHARE/DataBase/Ensembl/Homo_sapiens.GRCh38.pep.all.fa.sqldb
Use database:/hwfssz1/ST_CANCER/POL/SHARE/DataBase/Ensembl/Homo_sapiens.GRCh38.pep.all.fa
Load db file: /hwfssz1/ST_CANCER/POL/SHARE/DataBase/Ensembl/Homo_sapiens.GRCh38.pep.all.fa
Start indexing fasta file
10% 20% 30% 40% 50%Exception in thread "main" java.lang.ArrayIndexOutOfBoundsException: 268521
at com.compomics.util.experiment.identification.protein_inference.fm_index.Rank.getRank(Rank.java:145)
at com.compomics.util.experiment.identification.protein_inference.fm_index.WaveletTree.getRankRecursive(WaveletTree.java:376)
at com.compomics.util.experiment.identification.protein_inference.fm_index.WaveletTree.getRankRecursive(WaveletTree.java:379)
at com.compomics.util.experiment.identification.protein_inference.fm_index.WaveletTree.getRank(WaveletTree.java:358)
at com.compomics.util.experiment.identification.protein_inference.fm_index.WaveletTree.prepareWaveletTree(WaveletTree.java:212)
at com.compomics.util.experiment.identification.protein_inference.fm_index.WaveletTree.<init>(WaveletTree.java:157)
at com.compomics.util.experiment.identification.protein_inference.fm_index.FMIndex.addDataToIndex(FMIndex.java:1335)
at com.compomics.util.experiment.identification.protein_inference.fm_index.FMIndex.init(FMIndex.java:1118)
at com.compomics.util.experiment.identification.protein_inference.fm_index.FMIndex.<init>(FMIndex.java:544)
at main.java.pg.PepMapping.init(PepMapping.java:82)
at main.java.pg.PepMapping.loadDB(PepMapping.java:48)
at main.java.pg.InputProcessor.searchRefDB(InputProcessor.java:400)
at main.java.pg.PeptideSearchMT.main(PeptideSearchMT.java:332)
Hello. I'm interested in importing PepQuery into the Galaxy platform. Are you planning on putting the source code on here soon? I'd like to get a Conda recipe going.
I ran the stand-alone version of PepQuery on Linux and used this command:
java -jar pepquery.jar -pep peptide.txt -db custom1.fasta -ms HLA-1.mgf -varMod 21,117 -maxVar 5 -tol 20 -tolu ppm -e 0 -cpu 4 -minLength 8 -maxLength 15
And it said all the peptides on the file were invalid.
I then added the peptide used in the demonstration of the web application in the document, removing the ending R to simulate the peptides I have. PepQuery only recognised that peptide as valid. And even a random peptide is treated as valid (although it did not match the MGF file well.
How could I solve the issue and what would be the reason for it? Thanks
Hello Bo,
I have stumbled over one observation, which makes it difficult the output interpretation. I run pepquery search with enabled "-um -hc FALSE" options. Then I check my output for certain peptide, let's say "MPPSGSQAPPGSQSIMMSWMPPLAPCAACPCSPAPTLCPAHPAR", and I get in the sample A: score = "12.2598876", p-value="0.002997" and n_ptm="16", while in the sample B: score = "13.8024207", p-value = "9.99E-04" and n_ptm="0". Eyeballing the MS/MS spectra suggests that both results are highly similar. However, based on the output, I should drop the peptide based on the sample A, and retain it based on the sample B. So, my question is: this peptide - false or true positive?
Also, second observation: I am getting multiple results with n_db=0, p_value < 0.01 and n_ptm="-1". Could you help me to understand, what these hits mean?
Thank you,
Vladimir
javax.net.ssl.SSLException: Connection reset
at sun.security.ssl.Alert.createSSLException(Alert.java:127)
at sun.security.ssl.TransportContext.fatal(TransportContext.java:324)
at sun.security.ssl.TransportContext.fatal(TransportContext.java:267)
at sun.security.ssl.TransportContext.fatal(TransportContext.java:262)
at sun.security.ssl.SSLTransport.decode(SSLTransport.java:138)
at sun.security.ssl.SSLSocketImpl.decode(SSLSocketImpl.java:1400)
at sun.security.ssl.SSLSocketImpl.readApplicationRecord(SSLSocketImpl.java:1368)
at sun.security.ssl.SSLSocketImpl.access$300(SSLSocketImpl.java:73)
at sun.security.ssl.SSLSocketImpl$AppInputStream.read(SSLSocketImpl.java:962)
at org.apache.http.impl.io.SessionInputBufferImpl.streamRead(SessionInputBufferImpl.java:137)
at org.apache.http.impl.io.SessionInputBufferImpl.fillBuffer(SessionInputBufferImpl.java:153)
at org.apache.http.impl.io.SessionInputBufferImpl.read(SessionInputBufferImpl.java:205)
at org.apache.http.impl.io.ContentLengthInputStream.read(ContentLengthInputStream.java:176)
at org.apache.http.conn.EofSensorInputStream.read(EofSensorInputStream.java:135)
at com.amazonaws.internal.SdkFilterInputStream.read(SdkFilterInputStream.java:90)
at com.amazonaws.event.ProgressInputStream.read(ProgressInputStream.java:180)
at com.amazonaws.internal.SdkFilterInputStream.read(SdkFilterInputStream.java:90)
at com.amazonaws.internal.SdkFilterInputStream.read(SdkFilterInputStream.java:90)
at com.amazonaws.internal.SdkFilterInputStream.read(SdkFilterInputStream.java:90)
at com.amazonaws.event.ProgressInputStream.read(ProgressInputStream.java:180)
at java.security.DigestInputStream.read(DigestInputStream.java:161)
at com.amazonaws.services.s3.internal.DigestValidationInputStream.read(DigestValidationInputStream.java:59)
at com.amazonaws.internal.SdkFilterInputStream.read(SdkFilterInputStream.java:90)
at com.amazonaws.services.s3.internal.S3AbortableInputStream.read(S3AbortableInputStream.java:125)
at com.amazonaws.internal.SdkFilterInputStream.read(SdkFilterInputStream.java:90)
at java.util.zip.InflaterInputStream.fill(InflaterInputStream.java:238)
at java.util.zip.InflaterInputStream.read(InflaterInputStream.java:158)
at java.util.zip.GZIPInputStream.read(GZIPInputStream.java:117)
at sun.nio.cs.StreamDecoder.readBytes(StreamDecoder.java:284)
at sun.nio.cs.StreamDecoder.implRead(StreamDecoder.java:326)
at sun.nio.cs.StreamDecoder.read(StreamDecoder.java:178)
at java.io.InputStreamReader.read(InputStreamReader.java:184)
at java.io.BufferedReader.fill(BufferedReader.java:161)
at java.io.BufferedReader.readLine(BufferedReader.java:324)
at java.io.BufferedReader.readLine(BufferedReader.java:389)
at main.java.pg.S3Interface.downloadGZip(S3Interface.java:126)
at main.java.pg.S3Interface.download(S3Interface.java:95)
at main.java.msio.IndexFileDownloadWorker.download(IndexFileDownloadWorker.java:34)
at main.java.msio.IndexFileDownloadWorker.run(IndexFileDownloadWorker.java:23)
at java.util.concurrent.ThreadPoolExecutor.runWorker(ThreadPoolExecutor.java:1149)
at java.util.concurrent.ThreadPoolExecutor$Worker.run(ThreadPoolExecutor.java:624)
at java.lang.Thread.run(Thread.java:750)
Suppressed: java.net.SocketException: Broken pipe (Write failed)
at java.net.SocketOutputStream.socketWrite0(Native Method)
at java.net.SocketOutputStream.socketWrite(SocketOutputStream.java:111)
at java.net.SocketOutputStream.write(SocketOutputStream.java:155)
at sun.security.ssl.SSLSocketOutputRecord.encodeAlert(SSLSocketOutputRecord.java:81)
at sun.security.ssl.TransportContext.fatal(TransportContext.java:355)
... 40 more
Caused by: java.net.SocketException: Connection reset
at java.net.SocketInputStream.read(SocketInputStream.java:210)
at java.net.SocketInputStream.read(SocketInputStream.java:141)
at sun.security.ssl.SSLSocketInputRecord.read(SSLSocketInputRecord.java:464)
at sun.security.ssl.SSLSocketInputRecord.decodeInputRecord(SSLSocketInputRecord.java:237)
at sun.security.ssl.SSLSocketInputRecord.decode(SSLSocketInputRecord.java:190)
at sun.security.ssl.SSLTransport.decode(SSLTransport.java:109)
... 37 more
Dear PepQuery2 developers,
I entered a .txt file, but the following error appears:
Exception in thread "main" java.lang.IllegalArgumentException: No amino acid found for letter ..
at com.compomics.util.experiment.biology.aminoacids.AminoAcid.getAminoAcid(AminoAcid.java:274)
at com.compomics.util.experiment.identification.protein_inference.fm_index.FMIndex.createPeptideCombinations(FMIndex.java:1645)
at com.compomics.util.experiment.identification.protein_inference.fm_index.FMIndex.getProteinMappingWithoutVariants(FMIndex.java:1819)
at com.compomics.util.experiment.identification.protein_inference.fm_index.FMIndex.getProteinMapping(FMIndex.java:1796)
at main.java.pg.PepMapping.hasProtein(PepMapping.java:101)
at main.java.pg.InputProcessor.searchRefDB(InputProcessor.java:421)
at main.java.pg.PeptideSearchMT.search(PeptideSearchMT.java:337)
at main.java.pg.PeptideSearchMT.search_multiple_datasets(PeptideSearchMT.java:738)
at main.java.pg.PeptideSearchMT.main(PeptideSearchMT.java:183)
at main.java.pg.IMain.main(IMain.java:30)
My file is in this format(more than 1000 rows):
MRDPVSSQYSSFLFWRMPIPELDLSELEGLGLSDTATYKVKDSSVGKMIGQATAADQEKNPEGDGLLEYSTFNFWRAPIASIHSFELDLL
MYQGFWSEADVTRPFVSQAVITDGKYFSFFCYQLNTLALTTQADQNNPRKNICWGTQSKPLYETIEDNDVKGFNDDVLLQIVHFLLNRPKEEKSQLLEN
MNAIVALCHFCELHGPRTLFCTEVLHAPLPQGDGNEDSPGQGEQAEEEEAASLQVPPAHEAGPGCPFFPHAGLARSHGEPGDLEKQRRGPRPVSF
Thanks for your help!
Hello,
what is exact utility of "total_db" output parameter? If my predicted PSM has "n_db" = 0 (which means, that this spectrum ddi not find anything in reference db as a better match defined by score), so then why do I have non-zero values in "total_db"? Should I worry about high (>100) numbers in this field?
Also, if predicted "charge" of the peptide is higher then 3 - is it devaluates my PSM hit or not?
Thank you!
Vladimir
Dear Team I would reanalyze CPTAC data (endometrial PNNL files ) for some peptides of my interest. I'm new to proteomic analysis :-).
I downloaded 16 PNNL from the cptac site. Each PNNL is the combination of 10 TMT plexes with different samples from different patients pulled together. I'm able to analyze the raw files (after converting them in mgf with msconvert) and for each file i extract a list of significant peptides in the pool but I would like to know also the intensity levels of the peptides from each different sample. Therefore I suppose that I should associate to the found peptides, the “TMT reporter ion” intensity from the different TMT channels, e.g., 127C, 128N,
Is there a way to achieve this goal with pepquery?
Here it is my code
java -Xmx10G -jar /databis/pepquery-1.6.2/pepquery-1.6.2.jar -o ‘‘pep’’ -fixMod 6,62,108 -varMod 117 -tol 10 -tolu ppm -um -itol 0.05 -prefix ‘‘pep’’ -t 1 -ms 01CPTAC_UCEC_W_PNNL_20170922_B1S1_f01.mgf -i "extended10FSP.fa" -db "/data/PIPELINE/GENOMES/hg38/TRANSLATECDS2.0/sequence.fasta" -n 1000 -m 1 -maxLength 50 -minLength 5 -um -hc FALSE -cpu 10
Hi,
I have custom MGF files from an immunopeptidomics experiement and a set of predicted neopeptides. I have tried running Pepquery to analyze these data with the following command (as an example):
java -jar /net/pepquery-1.6.2.jar -o /net/pep_output -varMod 75,117 -e 0 -tol 20 -tolu ppm -itol 0.05 -prefix pep_true -ms /net/combined.mgf -pep EEISNLKAAF -db /net/TEST_REF.fasta -n 1000 -m 1 -maxLength 11 -minLength 8 -um -hc FALSE
This command does not return most of the expected outputs (e.g., psm_rank.txt). I have run the command with some 'positive control' self peptides that were detected via Mascot, etc by some colleagues, but even these fail to produce any PSMs (including after removing these peptides from the reference fasta file). This suggests to me that the issue has to do with my parameters or perhaps input preparation. Do you have any suggestions? Is Open-pFind a prerequisite to running Pepquery on immunopeptidomics data? (I use a Mac)
All outputs of this command look something like this:
#############################################
PepQuery parameter:
Fixed modification: 6 = Carbamidomethylation of C
Variable modification: 75,117 = Acetylation of protein N-term,Oxidation of M
Max allowed variable modification: 3
Add AA substitution: false
Enzyme: 0
Max Missed cleavages: 100
Precursor mass tolerance: 20.0
Precursor ion mass tolerance unit: ppm
Fragment ion mass tolerance: 0.05
Fragment ion mass tolerance unit: Da
Scoring algorithm: 1 = Hyperscore
Min score: 5.0
Min peaks: 10
Min peptide length: 8
Max peptide length: 11
Min peptide mass: 500.0
Max peptide mass: 10000.0
Random peptide number: 1000
CPU: 32
#############################################
Input peptide sequences:1
Don't find indexed database:/net/TEST_REF.fasta.sqldb
Use database:/net/TEST_REF.fasta
Load db file: /net/TEST_REF.fasta
Start indexing fasta file
Indexing took 0.822619151 seconds and consumes 31.37724 MB
2021-07-20 21:54:27 [ INFO] Valid target peptides: 1
2021-07-20 21:54:27 [ INFO] Generate peptide objects ...
CPU: 1
Exception in thread "pool-1-thread-1" 2021-07-20 21:54:27 [ INFO] Generate peptide objects done.
java.lang.NullPointerException
at com.compomics.util.experiment.identification.utils.ModificationUtils.getPossibleModificationSites(ModificationUtils.java:197)
at main.java.pg.PeptideInput.calcPeptideIsoforms(PeptideInput.java:135)
Build target peptide index done. at main.java.pg.CreatePeptideInputWorker.run(CreatePeptideInputWorker.java:29)
at java.util.concurrent.ThreadPoolExecutor.runWorker(ThreadPoolExecutor.java:1142)
at java.util.concurrent.ThreadPoolExecutor$Worker.run(ThreadPoolExecutor.java:617)
at java.lang.Thread.run(Thread.java:748)
Finished:10000
Finished:20000
Finished:30000
Finished:40000
Matched spectra: 0
Matched PSMs: 0
Finished:10000
Finished:20000
Finished:30000
Finished:40000
Saved spectra: 0
Get matched spectra done!
2021-07-20 21:55:05 [ INFO] Time elapsed: 0.65 min
Don't find indexed database:/net/TEST_REF.fasta.sqldb
Use database:/net/TEST_REF.fasta
Use enzyme:NoEnzyme
Use enzyme:NoEnzyme
Enzyme: NoEnzyme, maxMissedCleavages: 100
Exception in thread "main" java.lang.NullPointerException
at com.compomics.util.experiment.identification.utils.ModificationUtils.getPossibleModificationSites(ModificationUtils.java:197)
at main.java.pg.PeptideInput.calcPeptideIsoforms(PeptideInput.java:135)
at main.java.pg.DatabaseInput.readDB(DatabaseInput.java:383)
at main.java.pg.PeptideSearchMT.main(PeptideSearchMT.java:542)
I ran pepquery stand-alone version and got this error (same in linux and windows system).
How can I resolve this problem?
Exception in thread "main" java.sql.SQLException: The database has been closed
at org.sqlite.core.NativeDB.throwex(NativeDB.java:471)
at org.sqlite.core.NativeDB.errmsg_utf8(Native Method)
at org.sqlite.core.NativeDB.errmsg(NativeDB.java:137)
at org.sqlite.core.DB.newSQLException(DB.java:921)
at org.sqlite.core.DB.throwex(DB.java:886)
at org.sqlite.core.NativeDB._open_utf8(Native Method)
at org.sqlite.core.NativeDB._open(NativeDB.java:71)
at org.sqlite.core.DB.open(DB.java:174)
at org.sqlite.core.CoreConnection.open(CoreConnection.java:220)
at org.sqlite.core.CoreConnection.(CoreConnection.java:76)
at org.sqlite.jdbc3.JDBC3Connection.(JDBC3Connection.java:26)
at org.sqlite.jdbc4.JDBC4Connection.(JDBC4Connection.java:24)
at org.sqlite.SQLiteConnection.(SQLiteConnection.java:45)
at org.sqlite.JDBC.createConnection(JDBC.java:114)
at org.sqlite.JDBC.connect(JDBC.java:88)
at java.sql.DriverManager.getConnection(DriverManager.java:664)
at java.sql.DriverManager.getConnection(DriverManager.java:270)
at main.java.pg.SpectraInput.readMSMSdb(SpectraInput.java:358)
at main.java.pg.PeptideSearchMT.main(PeptideSearchMT.java:336)
Exception in thread "pool-84-thread-1" com.amazonaws.SdkClientException: Unable to execute HTTP request: The target server failed to respond
at com.amazonaws.http.AmazonHttpClient$RequestExecutor.handleRetryableException(AmazonHttpClient.java:1219)
at com.amazonaws.http.AmazonHttpClient$RequestExecutor.executeHelper(AmazonHttpClient.java:1165)
at com.amazonaws.http.AmazonHttpClient$RequestExecutor.doExecute(AmazonHttpClient.java:814)
at com.amazonaws.http.AmazonHttpClient$RequestExecutor.executeWithTimer(AmazonHttpClient.java:781)
at com.amazonaws.http.AmazonHttpClient$RequestExecutor.execute(AmazonHttpClient.java:755)
at com.amazonaws.http.AmazonHttpClient$RequestExecutor.access$500(AmazonHttpClient.java:715)
at com.amazonaws.http.AmazonHttpClient$RequestExecutionBuilderImpl.execute(AmazonHttpClient.java:697)
at com.amazonaws.http.AmazonHttpClient.execute(AmazonHttpClient.java:561)
at com.amazonaws.http.AmazonHttpClient.execute(AmazonHttpClient.java:541)
at com.amazonaws.services.s3.AmazonS3Client.invoke(AmazonS3Client.java:5456)
at com.amazonaws.services.s3.AmazonS3Client.invoke(AmazonS3Client.java:5403)
at com.amazonaws.services.s3.AmazonS3Client.getObjectMetadata(AmazonS3Client.java:1372)
at com.amazonaws.services.s3.AmazonS3Client.getObjectMetadata(AmazonS3Client.java:1346)
at com.amazonaws.services.s3.AmazonS3Client.doesObjectExist(AmazonS3Client.java:1427)
at main.java.pg.S3Interface.download(S3Interface.java:54)
at main.java.msio.IndexFileDownloadWorker.download(IndexFileDownloadWorker.java:34)
at main.java.msio.IndexFileDownloadWorker.run(IndexFileDownloadWorker.java:23)
at java.util.concurrent.ThreadPoolExecutor.runWorker(ThreadPoolExecutor.java:1149)
at java.util.concurrent.ThreadPoolExecutor$Worker.run(ThreadPoolExecutor.java:624)
at java.lang.Thread.run(Thread.java:750)
Dear:
I was recently using pep to process cptac data, but the following error was reported
2021-11-04 15:14:32 [ INFO] Generate peptide objects ...
2021-11-04 15:14:32 [ INFO] Generate peptide objects done.
Exception in thread "main" org.sqlite.SQLiteException: [SQLITE_ERROR] SQL error or missing database (no such table: msmsdb)
at org.sqlite.core.DB.newSQLException(DB.java:909)
at org.sqlite.core.DB.newSQLException(DB.java:921)
at org.sqlite.core.DB.throwex(DB.java:886)
at org.sqlite.core.NativeDB.prepare_utf8(Native Method)
at org.sqlite.core.NativeDB.prepare(NativeDB.java:127)
at org.sqlite.core.DB.prepare(DB.java:227)
at org.sqlite.core.CorePreparedStatement.(CorePreparedStatement.java:41)
at org.sqlite.jdbc3.JDBC3PreparedStatement.(JDBC3PreparedStatement.java:30)
at org.sqlite.jdbc4.JDBC4PreparedStatement.(JDBC4PreparedStatement.java:19)
at org.sqlite.jdbc4.JDBC4Connection.prepareStatement(JDBC4Connection.java:48)
at org.sqlite.jdbc3.JDBC3Connection.prepareStatement(JDBC3Connection.java:263)
at org.sqlite.jdbc3.JDBC3Connection.prepareStatement(JDBC3Connection.java:235)
at main.java.pg.SpectraInput.readMSMSdb2(SpectraInput.java:319)
at main.java.pg.PeptideSearchMT.main(PeptideSearchMT.java:451)
my command is:
java -cp pepquery-1.6.2.jar main.java.index.BuildMSlibrary -c 12 -i TCGA-2014/ -o test_all
java -Xmx24G -jar pepquery-1.6.2.jar -o out2 -fixMod 6 -varMod 117 -tol 20 -tolu ppm -um -itol 0.5 -prefix pepout -ms test_all -pep input.fa -n 1000 -db uniprot.fa -maxLength 50 -minLength 5 -maxVar 5 -um -cpu 12 -indexType 1
How can I resolve the error? Looking forward to the reply
Hi,
I can not download the example dataset (iPRG-2015) for the stand-alone application as well as the procedures (Procedures_for_using_PepQuery_01232018.pdf). Please check the error message as follows.
Forbidden
You don't have permission to access this resource.
We have followed the procedure in: http://pepquery.org/data/PepQuery_for_immunopeptidomics_data.pdf for evaluating potential neoantigens using no-enzyme.
PepQuery2 does not have option: -tag
Does the the index function in PepQuery2 replace the utility of the tags file?
This is a question about how PepQuery handles isobaric substitutions. We search two different spectra files for the same peptide with the same protein reference database. PepQuery indicates that a spectrum from on of the files fits better to our peptide than any proteins in the reference DB with unrestricted modification search. However in the other file, only a spectrum scored higher on a protein in the reference DB with a deamidation of N (isobaric to D) is assigned.
We expected to see the PSM with the modified reference peptide in the first analysis as well but it doesn't show.
The Genome Reasearch Publication clearly describes how the scores are calculated but I have trouble understanding how the score between an isobaric PTM peptide and the queried peptide are different. Since the deamidation of N is the exact mass of D should both PSMs not be assigned the same score?
My mgf files with PepQuery results can be downloaded from here:
https://drive.google.com/file/d/145Ne9qpGQTu_fpFg0jXiS3-DVHXDpJm6/view?usp=sharing
(Fr4S31 is the one where our peptide is most confident and Fr2S31 is the one where a better matched N[deamidated]/D reference protein)
thank you!
Hello,
I am testing standalone pepquery on Linux machine through running several commands with different parameters. I noticed some inconsistencies in the output and strange warnings in stderr.
Machine configuration:
OS: Ubuntu 18.04.3 LTS (Bionic Beaver)
Linux kernel: 4.15.0-64-generic
Java: openjdk 11.0.4 2019-07-16, OpenJDK 64-Bit Server VM (build 11.0.4+11-post-Ubuntu-1ubuntu218.04.3, mixed mode, sharing)
Each command uses one *.mgf file stored in ${fbn}. list of peptides is stored in file $PEPTIDE. Indexed human proteome is stored in $PDB. Here are the three commands:
when I run ".1" and ".2", I get the following in stderr:
SLF4J: Class path contains multiple SLF4J bindings.
SLF4J: Found binding in [jar:file:/mnt/modules/pepquery/1.2.0/lib/slf4j-log4j12-1.6.1.jar!/org/slf4j/impl/StaticLoggerBinder.class]
SLF4J: Found binding in [jar:file:/mnt/modules/pepquery/1.2.0/lib/logback-classic-1.0.11.jar!/org/slf4j/impl/StaticLoggerBinder.class]
SLF4J: See http://www.slf4j.org/codes.html#multiple_bindings for an explanation.
SLF4J: Actual binding is of type [org.slf4j.impl.Log4jLoggerFactory]
WARNING: An illegal reflective access operation has occurred
WARNING: Illegal reflective access by com.sun.xml.bind.v2.runtime.reflect.opt.Injector$1 (file:/mnt/modules/pepquery/1.2.0/lib/jaxb-impl-2.2.4-1.jar) to method java.lang.ClassLoader.defineClass(java.lang.String,byte[],int,int)
WARNING: Please consider reporting this to the maintainers of com.sun.xml.bind.v2.runtime.reflect.opt.Injector$1
WARNING: Use --illegal-access=warn to enable warnings of further illegal reflective access operations
WARNING: All illegal access operations will be denied in a future release
"3" does not give any stderr.
Does this imply, that validation with unrestricted modification "-um" does not work?
Also, I have no difference between my output files ("psm_rank.txt") upon running "1", "2" or "3", which is a bit strange as "3" does not use "-um" at all.
Finally, in my "psm_rank,txt" I am getting many peptides with p-value = 100. Is it a bug?
Thank you.
Vladimir
no enzyme means cutting everywhere. Is there a No cut option? like the -pep?
Hi,
I have some questions about pepquery. I'm using pepquery find some peptides, which were searched using comet against Uniprot db. MS raw data set were downloaded from PRIDE. Only three data sets had results, and all the others had bugs. My parameter are:
Fixed modification: 6 = Carbamidomethylation of C
Variable modification: 21,114,117,118,128 = Acetylation of peptide N-term,Phosphorylation of Y,Oxidation of M,Phosphorylation of T,Phosphorylation of S
Max allowed variable modification: 5
Add AA substitution: false
Enzyme: 1
Max Missed cleavages: 2
Precursor mass tolerance: 20.0
Precursor ion mass tolerance unit: ppm
Fragment ion mass tolerance: 0.5
Fragment ion mass tolerance unit: Da
Scoring algorithm: 1 = Hyperscore
Min score: 12.0
Min peaks: 10
Min peptide length: 5
Max peptide length: 50
Min peptide mass: 500.0
Max peptide mass: 10000.0
Random peptide number: 1000
CPU: 32
First, I search 200 peptide with pepquery in a data set (32 mgf files), I met this:
pool-3-thread-13 -> Finished 137: LVQDIANNTNEEAGDGTTTATVLAR .
pool-3-thread-2 -> Finished 99: LVQDVASNTNEEAGDGTTTATVLAR .
Exception in thread "main" java.lang.ArrayIndexOutOfBoundsException: 14
at main.java.pg.RankPSM.rankPSMs(RankPSM.java:44)
at main.java.pg.PeptideSearchMT.main(PeptideSearchMT.java:682)
End Bye!
I search only 200 peptide with pepquery in next dataset(9 mgf file), there is a outofmemory bug. I give 110G memory, is that still not enough?
pool-3-thread-29 -> Finished 102: SAAEMYGSVTKHPSPSPLLSSSFDLDYDFQR .
pool-3-thread-7 -> Finished 33: KEEVEEDNEVSSGLKQNYDEMSPAGQISK .
pool-3-thread-22 -> Finished 152: PASLYQSSIDRSLERPMSSASMPSDFRK .
Exception in thread "main" java.lang.OutOfMemoryError
at java.lang.AbstractStringBuilder.hugeCapacity(AbstractStringBuilder.java:161)
at java.lang.AbstractStringBuilder.newCapacity(AbstractStringBuilder.java:155)
at java.lang.AbstractStringBuilder.ensureCapacityInternal(AbstractStringBuilder.java:125)
at java.lang.AbstractStringBuilder.append(AbstractStringBuilder.java:448)
at java.lang.StringBuilder.append(StringBuilder.java:136)
at java.lang.StringBuilder.append(StringBuilder.java:131)
at org.apache.commons.lang3.StringUtils.join(StringUtils.java:4132)
at org.apache.commons.lang3.StringUtils.join(StringUtils.java:4177)
at main.java.pg.PeptideSearchMT.main(PeptideSearchMT.java:643)
I search 200 peptide with pepquery in next dataset(31 mgf file), there is a kill bug. Why is that?
pool-3-thread-5 -> Finished 75: SIRGSKKPTNDSNPSR .
pool-3-thread-5 -> Start 79: ESCTEEIVSEAESHVSGISR ...
/var/spool/slurmd/job10720637/slurm_script: line 25: 25038 Killed java -Xmx110G -jar pepquery-1.6.2/pepquery-1.6.2.jar -o pride_out/${outdir} -cpu 32 -fixMod 6 -varMod 21,114,117,118,128 -tol 20 -tolu ppm -um -itol 0.5 -prefix pepqout -ms ${pepin}/${input} -pep ${fasta} -n 1000 -db 2020-11-26-reviewed-contam-UP000005640.fa -maxLength 50 -minLength 5 -maxVar 5 -indexType 1
Thank you very much in advance.
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.